

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Deoxycorticosterone acetate (DOCA)-salt hypertension in animals is an established model of volume-dependent hypertension with renal injury. It is induced in animals by the implantation of DOCA strips, which introduce high levels of mineralocorticoid that mimic aldosterone overload.1 Although mineralocorticoid is classically known to regulate renal tubular sodium reabsorption in the collecting duct, recent data indicate that it also causes inflammation and fibrosis in the kidney and cardiovascular system.2,3 It is well known that DOCA-salt hypertension is accompanied by renal injury, such as mesangial expansion, glomerulosclerosis, and tubulointerstitial fibrosis.4
The mammalian target of rapamycin (mTOR) is a ubiquitously expressed intracellular serine/threonine protein kinase that plays a crucial role in regulating cell proliferation and organ growth through effects on many cellular processes.5 Recently, it was suggested that activation of the mTOR pathway is involved in the glomerular hypertrophy associated with the loss of function in nephrons, interstitial fibrosis, and tubular atrophy that occur in several forms of kidney disease.6 In addition, the mineralocorticoid receptor-mediated activation of the mTOR signaling pathway results in cellular proliferation in human mesangial cells.7 However, the role of the mTOR signaling pathway in the pathogenesis of kidney injury in mineralocorticoid hypertension remains unclear. We therefore investigated the role of the mTOR signaling pathway in the pathogenesis of renal injury in DOCA-salt hypertension in this study.
MATERIALS AND METHODS
1. Animals
The use of animals in this study was approved by the Ethics Committee of Chonnam National University Medical School, and the experimental procedure conformed to the Institutional Guidelines for Experimental Animal Care and Use. Male Sprague-Dawley rats weighing 200 to 250 g were implanted with DOCA strips (200 mg/kg) 1 week following unilateral nephrectomy. Control rats were unilaterally nephrectomized without DOCA. Rats were then supplied with 0.9% saline to drink. Four weeks after DOCA implantation, systolic blood pressure (SBP) was measured by the tail-cuff method, and the rats were euthanized. Kidneys were then excised and processed for semiquantitative immunoblotting.
2. Semiquantitative immunoblotting
Whole kidneys were homogenized in ice-cold isolation solution, pH 7.2 (0.3 M sucrose, 25 mM imidazole, 1 mM EDTA, 8.5 µM leupeptin, 1 mM phenylmethylsulfonyl fluoride), and centrifuged at 1,000×g for 15 min at 4℃ to remove whole cells, nuclei, and mitochondria. The total protein concentration was measured by use of the Pierce BCA Protein Assay Reagent Kit (Pierce, Rockford, IL), and all samples were adjusted to the same final protein concentration with isolation solution. Samples were then solubilized in SDS-containing sample buffer for 15 min at 65℃ before they were stored at -20℃. SDS-PAGE was performed on 9% or 12% polyacrylamide gels by using the Bio-Rad Mini Protean II system (Bio-Rad, Hercules, CA, USA) and the proteins were transferred onto nitrocellulose membranes (Hybond ECL RPN3032D, Amersham Pharmacia Biotech, Little Chalfont, UK). Membranes were subsequently blocked with 5% milk in phosphate-buffered saline with Tween 20 (PBST), pH 7.5 (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaCl, 0.1% Tween 20), for 1 h. After an overnight incubation with 1:1000 dilutions of primary antibodies at 4℃, membranes were incubated with 1:1,500 dilutions of secondary anti-rabbit (P448, DAKO, Glostrup, Denmark) or anti-mouse (P447, DAKO, Glostrup, Denmark) horseradish peroxidase-conjugated antibodies. Proteins were visualized by using an enhanced chemiluminescence system.
3. Primary antibodies
Affinity-purified anti-rabbit antibodies against phosphorylated phosphatidylinositol-3-kinase (p-PI3K), phosphorylated Akt (p-Akt), phosphorylated mTOR (p-mTOR), caspase-3, Bax, and Bcl-2 were obtained from Cell Signaling Technology (Beverly, MA, USA). Other antibodies were obtained from the following suppliers: cyclooxygenase-2 (COX-2) antibodies were obtained from Cayman Chemical (Ann Arbor, MI, USA), transforming growth factor-β1 (TGF-β1) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-mouse antibodies against ED-1 were from Serotec (Raleigh, NC, USA), and α-smooth muscle actin (SMA) was from Sigma Chemical Co. (St. Louis, MO, USA).
RESULTS
1. Changes in functional parameters
Four weeks after DOCA implantation, SBP was significantly increased in DOCA-implanted rats compared with control rats (233±15 mmHg vs. 127±6 mmHg, respectively, p<0.05). The ratio of kidney weight to body weight was also increased (8.49±1.67 mg/g vs. 4.91±0.48 mg/g, p<0.05).
2. Effects of DOCA-salt hypertension on protein expression
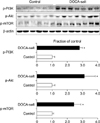
Fig. 1 and Table 1 depict changes in the phosphorylation state of mTOR signaling pathway proteins elicited by DOCA-salt hypertension. The expression levels of p-PI3K, p-Akt, and p-mTOR were significantly increased in the kidney in DOCA-salt hypertensive rats compared with the control rats. When the expression levels of inflammatory cytokines were examined, the expression of both ED-1 and COX-2 had increased in the kidney after treatment with DOCA-salt (Table 1). Likewise, the expression of TGF-β1, a pro-fibrotic molecule derived from inflammatory cells, and α-SMA, a molecular marker for myofibroblasts, was significantly increased in the kidney of DOCA-salt hypertensive rats (Fig. 2 and Table 1). Finally, the effects of DOCA-salt hypertension on apoptosis were examined by determining the expression levels of the pro-apoptotic molecular markers caspase-3 and Bax and the anti-apoptotic Bcl-2 protein. It was found that the expression of both pro-apoptotic proteins was significantly increased in the kidneys of DOCA-salt hypertensive rats compared with control rats, whereas the renal expression of Bcl-2 was decreased in rats after treatment with DOCA-salt (Fig. 3 and Table 1).
DISCUSSION
We have previously demonstrated that DOCA-salt-treated rats exhibit marked hypertension with impaired renal function, including decreased creatinine clearance, polyuria with decreased urine osmolality, renal hypertrophy, and proteinuria.8-10 In the present study, we showed that severe DOCA-salt hypertension also results in increased kidney weight.
It was previously shown that DOCA-salt treatment results in glomerulosclerosis and tubulointerstitial fibrosis, which is related to increased infiltration of inflammatory cells, increased expression of pro-inflammatory and pro-fibrotic molecules, and the epithelial-mesenchymal transition (EMT).9-11 When the expression of the inflammatory markers ED-1 and COX-2 was examined in the kidney in our rat model of DOCA-salt hypertension, it was found to be increased compared with that of control rats, as was the expression of TGF-β1 and α-SMA. TGF-β itself plays a crucial role in the pathogenesis of progressive renal disease, in which renal fibrosis is promoted by the accumulation of extracellular matrix. TGF-β may also induce the production of pro-apoptotic molecules such as caspase-3 and Bax, as well induce EMT.12,13 Our data showed that the renal expression of the pro-apoptotic proteins caspase-3 and Bax was increased and that of the anti-apoptotic factor Bcl-2 was decreased in the DOCA-salt model of hypertension. These changes may contribute to the renal injury that occurs in DOCA-salt hypertension; however, the upstream mechanism of these changes remains elusive.
It was previously demonstrated that activation of the mineralocorticoid receptor can stimulate the PI3K/Akt/mTOR pathway in human mesangial cells.7 Therefore, in this study, we used an established rat model in which DOCA-salt induces mineralocorticoid hypertension to investigate the changes that occur in the PI3K/Akt/mTOR signaling pathway in the kidney as a result of this state. This pathway leads to the phosphorylation and activation of mTOR, which plays a role in mediating cell size and mass, proliferation, and survival.14,15 In our study, PI3K, Akt, and mTOR proteins were all found to have increased phosphorylation levels in the kidneys of DOCA-salt hypertensive rats. Recent data have further revealed that the activation of the PI3K/Akt/mTOR pathway in the kidney results in glomerular hypertrophy, tubulointerstitial atrophy, and fibrosis in several animal models of kidney disease.5,6 Because renal hypertrophy may contribute to podocyte injury, proteinuria, and progressive loss of renal function, it is an important structural change that occurs in progressive renal disease.16 The activation of mTOR plays a pivotal role in the pathogenesis of renal hypertrophy in cases of compensatory renal hypertrophy and diabetic nephropathy.17-19 The activation of mTOR may also promote the tubular injury, atrophy, and interstitial fibrosis that occurs in response to interstitial inflammation and EMT following the stimulation of lymphocyte and fibroblast proliferation.20-22 In conclusion, our finding that the phosphorylation of PI3K/Akt/mTOR was increased in the kidneys of DOCA-salt hypertensive rats suggests that the mTOR signaling pathway is related to the pathogenesis of DOCA-salt hypertension with renal injury.



 XML Download
XML Download