

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Synapses in the brain are able to change their strength in response to neural activity and these modifications to synaptic strength are thought to contribute to memory formation and also to brain development. These modifications take the form of long-term potentiation (LTP), which is the activity-induced enhancement of synaptic transmission, and long term depression (LTD), which is the activity induced reduction in synaptic transmission. Whether LTP or LTD is induced is determined by the frequency of stimulation in the induction protocol, with LTP typically induced by high frequency stimulation and LTD typically induced by low frequency stimulation (LFS). In addition to these forms of plasticity, synapses that have recently undergone LTP can have this synaptic strengthening reversed in a process termed depotentiation (DP), a process that is also triggered by LFS.
Deficits in DP have recently been described as a result of manipulations that are thought to be relevant to schizophrenia, and so one of the aims of this review is to highlight the relevance that deficits in DP may have to this debilitating disorder. An additional question that will be addressed is whether DP and LTD constitute essentially the same phenomena, of if they are distinct processes. The question arises because both forms of plasticity can be induced by similar LFS type protocols, and because they result in a similar effect, which is a reduction in synaptic efficiency. As it is possible that LTP may have been induced previously at synapses being studied, due to the in vivo neuronal activity prior to acute slices being made, it remains a possibility that the only difference between DP and LTD is the time that has elapsed since a previous LTP inducing stimuli. If this is the case it seems to be unlikely that it will be possible to distinguish the role in behavior that these processes play, as it will not be possible to selectively antagonize one of them either pharmacologically or by genetic manipulation. If unique signaling pathways are uncovered however, these will serve to characterize DP as different from LTD and may allow the selective antagonism of DP allowing its role in behavior to be assessed. Also, if DP is dis-regulated in neurological diseases, understanding its molecular characteristics may provide pharmacological targets by which it may be modulated. This review will therefore also explore the molecular mechanisms involved in DP and highlight evidence that suggests that DP and LTD are in fact distinct processes and are relevant to different behaviors.
THE INDUCTION OF DP
DP is a form of synaptic plasticity that can be induced by two general patterns of synaptic stimulation, both in vitro and in vivo. The most widely studied induction protocol involves delivering LFS to the pathway being studied.1-4 Where LFS is used to induce this form of plasticity it is often delivered at a frequency that is shown to be ineffective at inducing LTD in the preparations used2,5 which is one indication that this phenomenon maybe of a distinct nature from LTD. Thus in some reports 2 Hz LFS is able to induce DP but is unable to induce LTD.2,5 The second pattern of stimulation that results in DP is theta frequency stimulation, either delivered after the induction of LTP,6-8 or during the LTP induction protocol itself.9 As theta oscillations are a characteristic of hippocampal neuronal activity10 induction protocols that are based on them may be said to be more physiologically relevant. In addition DP can be induced pharmacologically, by applying NMDA11 or DHPG12 at concentrations that are not able to induce LTD, as well as by application of the hormone leptin.13 These differences in the induction requirements for DP compared to LTD may indicate that after LTP synapses enter a different state compared to the basal state, in which they are more sensitive to specialized stimuli that are able to reduce the efficiency of synaptic transmission.
In some experimental conditions this form of plasticity has been observed to occur only within a short time frame after LTP induction.5,11,14 However in other conditions it has also been observed to occur outside this time frame.2,15,16 One possible explanation for these divergent findings is that the strength of the LTP induction protocol may determine how long after LTP the synapses remain susceptible to DP. For example where it has been observed that it is only possible to induce DP for a short time after LTP relatively strong LTP induction protocols are used. For example, repeated trains of high frequency stimulation, typically two 1 second tetani delivered at 100 Hz 20 seconds apart.5,11,14 However where DP is observed to occur more than 30 minutes after LTP induction, weaker protocols are used to induce LTP. These may be a single 100 Hz tetanus2 or theta bursts.15,16 Consistent with this hypothesis are the findings that additional signaling mechanisms are recruited if strong LTP induction protocols are employed, for example L-type voltage-dependent calcium channels.17,18 Potentially these additional signaling mechanisms may result in a form of LTP that becomes resistant to DP more quickly.
MOLECULAR MECHANISMS OF DP
One way to distinguish between DP and LTD is to examine the molecular mechanisms involved. In summary it appears that although some of the mechanisms in DP and LTD are held in common, evidence is accumulating that many molecular signaling pathways are distinct. For example DP is reported to be dependent on the activation of NMDA receptors in some experimental conditions3,11,14 and on metabotropic glutamate receptors in others2,19,20 which is similar to the case in LTD.21 However, although NMDA receptors are thought to play a role in the induction of both forms of plasticity, there is some evidence that the NMDA receptor subunit NR2B is involved in LTD whereas NR2A is involved in DP.22-24
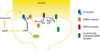
One of the first signaling pathways suggested to be involved in DP involves adenosine signaling. Exogenously applied adenosine is able to decrease the magnitude of LTP5,25 and antagonists of adenosine A1 receptors are able to block DP induced by synaptic stimulation5,26,27 however see.28 This mechanism is thought to be triggered by the efflux of cAMP and its extracellular conversion to adenosine, and to be via A1 receptors coupled to Gi/o proteins that reduce intracellular cAMP concentrations, thus abrogating PKA signaling.5 The current evidence is that adenosine receptors do not play a role in LTD in area CA1 of the hippocampus as, for example, LTD and LTP are both intact in A1 knock out mice29 and application of adenosine produces only a transient depression in synaptic transmission.30 The signaling induced by A1 receptor activation in DP is thought to converge on mechanisms involved in LTD however. Like in LTD,31 antagonists of protein phosphatase 1 (PP1) or protein phosphatase 2A (PP2A) block DP, possibly via the regulation of PKA signaling outlined above.5 In both LTD and DP it is suggested that protein phosphatases subsequently dephosphorylates Ca2+ calmodulin dependent protein kinase II (CaMKII), leading to the failure of LTP.32 There are differences in the requirements for protein phosphatases in DP and LTD however. For example if PP2A is selectively antagonized LTD is blocked but DP is retained.33 In addition a third protein phosphatase, Calcineurin Aα has been found to be specifically involved in DP, as in Calcineurin Aα knock out mice both LTP and LTD can be induced, whereas DP is absent.34
An additional down stream effector of adenosine A1 receptor activation in DP may be G-protein-activated inwardly rectifying K+ (GIRK) channels. These channels are stimulated by adenosine A1 receptors,35 and the trafficking of GIRK channels is regulated by similar mechanisms to those involved in DP, involving neuronal activity, NMDARs and PP1.36 Consistent with this, DP is deficient in GIRK2 knock out mice.35 As GIRK channels regulate neuronal excitability, this may suggest that the degree of excitability may be important for determining whether DP can occur, for example by regulating the magnitude of LTP that DP is reversing. Consistent with this, in animal models of disease in which excitability is enhanced, depotentiation is also absent.16 Please find these molecular mechanisms illustrated in Fig. 1.
Conversely several groups have found that targeted manipulations that block NMDA receptor dependent LTD, have no effect on DP. Firstly mice that are engineered to express the SV40 small t antigen known to inhibit PP2A, are deficient in NMDA dependent LTD, but exhibit LTP and DP of the same magnitude as controls.33 Secondly, mice that do not express a specific isoform of phosphatidylinositol-3-kinase (PI3K), PI3Kγ, also have a specific deficit in NMDA receptor dependent LTD, with intact LTP and DP.37 Thirdly, the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway has been found to be specifically involved in NMDA dependent LTD, but not in LTP or DP.38 In the first two cases behavioral experiments also showed a deficit in flexibility of memory, findings that firstly implicate NMDA receptor dependent LTD in this type of memory, and secondly indicate that DP is not required as it was intact in these models. Thus although a role has not yet been suggested for DP in a specific form of memory, by exclusion it appears that its role is not the same as for NMDA dependent LTD.
AMPA RECEPTOR TRAFFICKING IN DP
Similar to the expression of LTP and LTD39,40 AMPA receptor trafficking is involved in the expression of DP. AMPA receptors mediate the vast majority of excitatory neurotransmission in the mammalian brain, and their trafficking to and from synapses is under a high degree of regulation. AMPA receptor subunits have either long cytoplasmic tails (GluR1 and GluR2L) or short cytoplasmic tails (GluR2 and GluR3), and on these cytoplasmic tails are many binding sites that enable them to interact with specific binding partners to control their movements. In DP it is trafficking of AMPA receptor subunits that have long cytoplasmic tails that has been found to be particularly involved. This mechanism involves Rap2, c-Jun NH2-terminal kinase (JNK) and the NMDA receptor subunits NR2A.24 There is evidence that there are differences in the mechanisms of AMPA receptor trafficking in DP and LTD, however. For example in LTD AMPA receptor trafficking is not regulated by Rap2 and JNK, but by Rap1 and p38 MAPK dependent mechanism.24,41 In addition there are also distinct dephosphorylation patterns on AMPA receptors in LTD and DP. In LTD serine 845 of GluA1 is dephosphorylated, but not serine 831, and in DP the opposite arrangement is seen; serine 831 is dephosphorylated and serine 845 is not.15 Thus although signaling mechanism involved in DP and LTD may converge on the same expression mechanism, AMPA receptor trafficking, the regulation is demonstrably different, again pointing to distinct forms of plasticity.
THE RELEVANCE OF DP TO SCHIZOPHRENIA
One theory on the etiology of Schizophrenia suggests that it could be caused by subtle differences in neurodevelopment that result in erroneous neuronal connectivity.42 A range of factors influences the chance of developing Schizophrenia including genetic factors and environmental insults during development. Several specific genes have been linked to this disease, and so it is possible that alterations in their function may contribute to its symptoms. One such gene is Neuregulin 1 (NRG-1).43 It is therefore of note that a role has also been discovered for NRG-1 in DP, via activation of D4 dopamine receptors.44,45 This finding suggests that a deficit in this form of synaptic plasticity may play a role in the etiology of this disease. Also, ErbB4, the receptor for NRG-1, is expressed on parvalbumin (PV) expressing interneurons,46 which is of interest as there are consistent physiological deficits reported to be present in schizophrenia, one of which is disruption to GABAergic inhibition, as evidenced by a loss of the GABA synthesizing enzyme glutamic acid decarboxylase-67 (GAD67).47 Importantly the loss of GAD67 in the hippocampus occurs preferentially in PV expressing interneurons and the expression of PV itself in these interneurons is also reduced in Schizophrenic patients.48 Therefore the recent finding that DP is absent in mice in which the expression of ErbB4 has been specifically ablated in PV expressing interneurons,49 provides a convincing case that NRG-1 function is necessary for DP through an effect on the neuronal network, and that in schizophrenia a deficit in DP may result in behavioral or cognitive abnormalities.
Despite the reported genetic associations in schizophrenia, a clear pathophysiological role has not been established for them all, and it is unlikely that one genetic deficit alone could underlie the plethora of symptoms of this disease. In addition the rate of concordance for Schizophrenia between twins is only 50%.50 Among the various environmental insults that have been implicated as risk factors emphasis has been given to maternal starvation, perinatal infections, and cannabis use.51 In order to study the role of neurodevelopmental disruption in Schizophrenia in a controlled way, the effect of a single injection with the methylating agent methylazoxymethanol acetate (MAM) on embryonic day 17 has been studied. This disrupts development in a temporally specific way, and results in behavioral and histopathological changes that are thought to be similar to those observed in schizophrenia.52 Interestingly there is also a deficit in DP in this model of schizophrenia, although LTP and LTD remain intact.16 That a less specific molecular manipulation is able to result in a similar deficit to the highly specific ablation of NRG-1 signaling adds further weight to the hypothesis that risk factors of different natures may converge on common physiological deficiencies, like a deficit in DP, and that these common abnormalities may then underlie the symptoms observed in schizophrenia.
POSSIBLE IMPLICATIONS OF A DEFICIT IN DP
The role of DP is not currently known. It has been suggested however that during development, DP prevents spurious information being stored that may otherwise be encoded by the incidental correlated firing of neurons.53 More generally, this suggests that depotentiation may act as a quality control mechanism for information storage and it is tempting to speculate that a deficit in depotentiation may have a detrimental effect on the verisimilitude of the information that is encoded by the brain. In the context of Schizophrenia therefore, a deficit in depotentiation may, by allowing uncorroborated information to be stored, contribute to the positive symptoms experienced by patients of this disease. Recent evidence that is consistent with this hypothesis is that mice in which ErbB4 expression has been ablated in PV interneurons, and that show a deficit in DP, also show behavioral deficits that are thought to resemble the positive symptoms of schizophrenia.49 This hypothesis will be difficult to prove, however if correct it would suggest that tailoring pharmacological therapies to enhance DP might have benefits for Schizophrenic patients.
 XML Download
XML Download