

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pure red cell aplasia (PRCA) is a disorder that was first described in 1922 and that is characterized as a normocytic anemia associated with the absence of erythroblasts in the bone marrow. PRCA can be induced by various causes.1 However, only a few cases of PRCA associated with hepatitis A have been reported.2-6 In Korea, there have been several reports of PRCA associated with thymoma, lymphoma, SLE, Hashimoto's disease, parvovirus, and adverse drug reactions. However, no case of PRCA caused by acute hepatitis A has yet been reported. In this report, we describe a case of PRCA with acute hepatitis A and review the literature.
CASE REPORT
A 36-year-old male was admitted with a 3-day history of fever, jaundice, and vomiting. His medical history was unremarkable. On admission, he was alert and oriented. His vital signs were as follows: blood pressure, 120/90 mmHg; heart rate, 88 beats/min; respiratory rate, 18/min; and body temperature, 37.1℃. He had obvious jaundice and icteric sclera. The conjunctiva was not anemic. The liver and spleen were not palpable. His hemoglobin concentration was 17.1 g/dL, his red cell count was 5.74×106/µL, his white blood cell count was 5,000/µL, and his platelet count was 114,000/µL. Blood chemistry analyses revealed a total bilirubin level of 3.8 mg/dL; direct bilirubin, 2.39 mg/dL; AST, 7736 IU/L; ALT, 3558 IU/L; LDH, 14,407 IU/L; albumin, 3.8 g/dL; prothrombin time, 18.6 s (10.1-12.4 s); BUN, 50.9 mg/dL; and Cr, 5.7 mg/dL.
The test for hepatitis A virus IgM antigen was positive. Tests for hepatitis B surface antigen, and IgM anti-hepatitis B core, and anti-hepatitis C virus antibody were all negative. Tests for human immunodeficiency virus antibody and Epstein-Barr virus VCA IgM antibody were negative and parvovirus was undetectable by polymerase chain reaction. Tests for anti-nuclear antibody and rheumatoid arthritis factor were negative. C3 was 86.6 mg/dL, C4 was 53 mg/dL, IgG was 746 mg/dL, IgA was 442 mg/dL, and IgM was 469 mg/dL. Direct Coombs' test was positive and haptoglobin decreased to 3.5 mg/dL. The results of a chest X-ray, abdominal ultrasonography, and duodenoscopy were unremarkable.
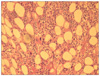
He received hemodialysis treatment owing to acute renal failure three times a week from day 2. On day 4, he was diagnosed with hepatitis A and treated with fluid therapy. On admission, there was no sign of anemia, but his hemoglobin level had gradually decreased. No sign of gastrointestinal bleeding was observed. On day 23, his hemoglobin level had dropped to 4.8 g/dL and his reticulocyte count had significantly decreased to 0.13%. The peripheral blood smear showed microcytic, normochromic anemia with anisopoikilocytosis. A total of 6 U of red cells were administered for 3 days. Bone marrow biopsy and aspiration were performed on day 25. The bone marrow biopsy showed 50% cellularity and the myeloid:erythroid ratio was 16.1:1. Marked hypoplastic erythropoiesis was observed (Fig. 1). A paucity of erythroid precursors was noted, although granulopoiesis was normal in number and maturation. Megakaryocytes were normally observed (Fig. 2). Differential erythroid cell counts of aspirates were as follows: pronormoblast, 1.4%; basophilic normoblast, 2.4%; polychromatic normoblast, 0.0%; and orthochromatic normoblast, 0.0%.
As a result of these findings, he was diagnosed with pure red cell aplasia associated with acute hepatitis A. He began treatment with intravenous methylprednisolone (1 mg/kg/day) daily at day 29. After the steroid therapy, his hemoglobin level and renal function began to improve steadily. Hemodialysis was stopped on day 34, and the hemoglobin level had increased to 8.3 g/dL on discharge. Methylprednisolone was tapered by 10 mg every week until discharge. One month after discharge, the prednisolone dose was tapered to 10 mg in the outpatient clinic. On day 48 after discharge, prednisolone treatment was stopped, because the hemoglobin level had improved to 13.9 g/dL and the Cr level had decreased to 1.3 mg/dL (Fig. 3).
DISCUSSION
PRCA results in an anemia associated with severe reticulocytopenia and the absence of marrow erythroid precursor cells.1 Innate PRCA is known as Blackfan-Diamond anemia, and is usually diagnosed among infants less than 1 year of age. In adults, PRCA usually occurs as an acquired disease, mainly due to thymoma, connective tissue disease, viral infection, lymphoma, and adverse drug reactions.1
In the present case, while the patient was being treated with fluid therapy and hemodialysis for hepatitis A with acute renal failure, his hemoglobin level gradually decreased. No signs of gastrointestinal bleeding were observed and no evidence of thymoma was apparent in the chest X-ray. Results were negative for human immunodeficiency virus antibody, Epstein-Barr virus VCA IgM antibody, viral hepatitis B antibody, viral hepatitis C antibody, anti-nuclear antibody, parvovirus PCR, and rheumatoid arthritis factor. There were no suspicious findings for lymphoma in the peripheral blood smear or bone marrow. His medical history was unremarkable. Bone marrow examination showed marked erythroid hypoplasia with normal granulocytes and megakaryocytes. As a result, he was diagnosed with PRCA caused by acute hepatitis A.
Globally, cases of acute hepatitis A associated with PRCA are very rare. In all of the cases reported to date, transient, severe anemia appeared after the acute phase of the hepatitis had passed and disappeared within a short period after blood transfusion or corticosteroid therapy. The clinical course of PRCA with hepatitis A and the prognosis of the patients were good.2-6
The mechanism of PRCA seen in many cases of viral hepatitis is poorly understood. Bone marrow and liver contain components of the reticuloendothelial system and may, therefore, be adversely affected by similar agents. Previously known main mechanisms are viral-induced autoimmune response and direct viral infection of bone marrow progenitor cells.1 The autoimmune response can directly cause a cytolytic reaction to bone marrow progenitor cells by T-cells, natural killer cells, or complement binding with antibody and can damage bone marrow stromal cells.1
Idiopathic PRCA can be initially treated with corticosteroids and blood transfusion. Generally, the recovery period is about 1 month.7 In a study of 27 PRCA patients using corticosteroids for 2 to 5 weeks, remission occurred within 4 weeks in 40% of cases, and a cure rate of 37% was reported.8 Case studies of PRCA with acute viral hepatitis are rare; cases of treatment with corticosteroids and blood transfusion have been reported.2-6
In corticosteroid-resistant cases, PRCA can be treated with cyclosporin A or cyclophosphamide. A study of 43 patients treated with cyclosporin A showed an overall response of 65%.9 One study reported that the duration of remission induced by cyclophosphamide seemed to be prolonged compared with that induced by corticosteroid.8 In addition, several studies reported that antithymocyte globulin, alemtuzumab, and rituximab are useful in the treatment of refractory PRCA.7
In summary, we have reported the first case of PRCA associated with acute hepatitis A in Korea. The case was treated with corticosteroids and transfusion, reflecting the previous literature. The patient showed an early response to corticosteroids and successfully achieved remission after 2 months of treatment.


 XML Download
XML Download