

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The first report of portal vein thrombosis (PVT) was made in 1868. That was a type of phlebothrombosis complicated with portal hypertension and a reduction of the blood flow to the liver. Owing to various symptoms in PVT, it is difficult to make an accurate diagnosis clinically.1 PVT causes portal hypertension and varix, and if severe, can cause variceal hemorrhage and ischemic bowel disease.1
The pathogenesis of PVT has not been identified clearly, but recent studies report that it is related to systemic causes such as protein C, protein S, and antithrombin III deficiency disorder.2 However, there are very few studies of optimal treatment owing to the low incidence rate (less than 0.004%).3
In Korea, there are several reports of acute PVT caused by oral contraceptives and pancreatitis, but few cases of PVT caused by a deficiency of anticoagulant factors and reperfused with anticoagulation therapy.4 We report a case of PVT caused by protein C and S deficiency that ended in successful reperfusion with anticoagulation therapy.
CASE REPORT
A 66-year-old man was admitted via the emergency department because of systemic myalgia and anorexia lasting for 2 weeks. He complained of high fever, nausea, and vomiting, and the results of liver function tests were abnormal.
He had taken antihypertensive medication for 5 years and clopidogrel for 4 years owing to a history of transient ischemic attack. He had no history of diabetes mellitus or liver diseases. He had undergone subtotal gastrectomy for early gastric cancer 2 years ago. On admission, his vital signs were as follows: blood pressure, 130/70 mmHg; resting heart rate, 70 beats per minute; respiratory rate, 20 breaths per minute; and body temperature, 37℃. His consciousness was clear but his conjunctivae were pale. His breath sounds were clear and his heart sounds were regular with no murmur. He did not complain of tenderness or rebound tenderness of the abdomen, and there were no signs of hepatomegaly or splenomegaly. The neurologic examination revealed no abnormal signs.
The laboratory findings were as follows: white blood cell count, 12,080/mm3 (normal: 4,000-10,000/mm3); hemoglobin, 10.5 g/dl (13.0-17.0 g/dl); hematocrit, 31.3% (39-52%); platelet count, 351,000/mm3 (150,000-40,000/mm3); serum aspartate aminotransferase (AST), 42 IU/L (0-37 IU/L); serum alanine aminotransferase (ALT), 137 (0-41 IU/L); serum alkaline phosphatase (ALP), 442 IU/L (39-117 IU/L); gamma(γ)-glutamyl transferase (γ-GT), 255 IU/L (11-50 IU/L); serum total bilirubin, 0.9 mg/dl (0.2-1.3 mg/dl); total protein, 7.3 g/dl (6.7-8.3 g/dl); albumin, 3.0 g/dl (3.5-5.2 g/dl); and prothrombin time international normalized ratio (PT INR), 1.21 (0.85-1.15). HBsAg, anti-HBs, and anti-HCV were negative.
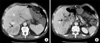
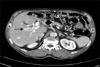
Abdominal CT scan showed complete occlusion of the intrahepatic portal vein by portal vein thrombi and small hepatic abscess around the occluded vessels (Fig. 1). Abdominal Doppler ultrasound, esophagogastroduodenoscopy, and coagulation factor analysis were performed to identify the cause of the PVT. The abdominal Doppler ultrasound showed complete portal vein occlusion but could not identify the surrounding microvessels, which implied a low probability of malignancy (Fig. 2A). Esophagogastroduodenoscopy showed no esophageal or gastric varices. Coagulant factor VIII and protein C factor were 261% (normal: 60-140%) and 41% (normal: 60-140%), respectively. Protein S factor showed a decrease of 14% (normal: 60-140%). Thus, the patient was diagnosed with PVT caused by a deficiency of protein C and S.
Administration of warfarin (at a dose of 5 mg twice daily) and subcutaneous injection of low molecular weight heparin (enoxaparin, 1 mg/kg, twice daily) was promptly started. At the time of discharge, the maintenance dose of warfarin was 2.5 mg per day (target INR: 2-2.5). The abdominal Doppler ultrasound, performed just before discharge, revealed that the portal vein was still completely occluded by thrombi but decreased in diameter. The results of the laboratory tests conducted on an outpatient basis were as follows: INR, 1.90; protein C, 41%; protein S, 14%; AST, 61 IU/L; ALT, 54 IU/L; ALP, 56 IU/L; and rGT, 215 IU/L. Abdominal Doppler ultrasound showed blood flow in the PVT (Fig. 2B). The results of the blood tests conducted 6 months later were as follows: serum INR, 1.71; AST, 57 IU/L; ALT, 46 IU/L; ALP, 113 IU/L; and γGT, 121 IU/L. On the abdominal Doppler ultrasound performed at that time, the portal vein thrombi had disappeared and a smooth bloodstream was observed in the portal vein (Fig. 2C, D, 3).
DISCUSSION
PVT interrupts blood flow to the liver and causes portal hypertension.3 Phlebothrombosis is caused by local, genetic, or acquired thrombophilic factors.5 Local factors include local inflammatory lesions such as neonatal funiculitis, diverticulitis, appendicitis, pancreatitis, duodenal ulceration, tuberculous lymphadenitis, portal vein injury by surgical operations, thrombokinesis, pressure mass effect by an abdominal neoplasm, and portal hypertension by liver cirrhosis.5 Denninger et al. reported that 6% to 11% of PVT is attributed to liver cirrhosis, and 35% is related to hepatocellular carcinoma.3 Webb et al. reported that 40% of PVT is caused by intraperitoneal or systemic septicemia. In other reports, 7% of PVT was reported to be caused by chronic pancreatitis.6 Primary myeloproliferative disorder, antiphospholipid antibody syndrome, paroxysmal nocturnal hemoglobinuria, oral contraceptives, pregnancy, childbirth, malignant tumor, and hypercysteinemia are reported as acquired thrombophilic factors.5 Genetic thrombophilic factors can be divided into the frequent type, such as factor V Leiden mutation and mutation IIG20210, in which PVT is rarely associated, and the rare type, such as protein C, S, and antithrombin III, in which PVT is more likely to occur.5
Clinically, PVT is classified as acute or chronic. However, it is difficult to identify when the symptoms begin and even the temporal criteria about chronicity have not yet been established.7 Malkowski et al. classified PVT as acute or chronic according to symptom onset time before admission (60 days).7 Acute PVT typically provokes stomachache, nausea, emesis, and fever. The symptoms can be more severe if mesenteric infarction provokes infarction of the bowel or liver. Nonspecific symptoms such as diarrhea, anorexia, weight loss, and abdominal distention may develop.8 The symptoms of chronic PVT are similar to the acute ones, but such symptoms may be related to portal hypertension and splenomegaly.9 Splenomegaly, anemia, leukopenia, thrombocytopenia, and variceal bleeding can be provoked.9
Our case did not develop portal hypertension-related complications but showed a fever, stomachache, nausea, and emesis, and these symptoms are compatible with PVT. Duplex, Doppler ultrasound, and abdominal CT scan can be used to make a accurate diagnosis of PVT in the early stage, and abdominal MRI can be another diagnostic tool.10 In particular, abdominal MRI is known to be more useful than Doppler ultrasound in identifying venous collateral development and cavernoma.10 In patients with PVT, local factors such as liver cirrhosis and pancreatitis should be suspected. If no local factor can be identified, it is necessary to investigate genetic or acquired thrombophilic factors.8
In our case, it was possible to infer that PVT was secondary to a liver abscess of the right lobe. However, it was more likely that the abscess was secondary to PVT for the following reasons. First, although massive thrombi obstructed the main and left portal vein, the abscess was restricted to the right lobe only. Second, it took more than 1 year after abscess resolution for the cholestatic features on the liver function tests to be normalized. Third, antibiotic therapy of only 3 weeks brought about a significant increase in main portal vein flow.
Regarding the treatment of PVT, long-term anticoagulant therapy and invasive methods such as surgical thrombectomy, local thrombolytic therapy, thrombolytic agentbased selective portography, and intrahepatic stent insertion via the jugular vein can be applied.9 In many cases, chronic PVT is accompanied by gastroesophageal varix, which can result in variceal bleeding during anticoagulation therapy.8 In the case of acute PVT, however, early anticoagulant therapy can prevent the spread of thrombi and promote blood reperfusion.9 Sheen et al. applied anticoagulant therapy to 9 acute PVT patients with a target INR of 2 to 4 for 3 months. They reported that reperfusion was achieved in 78%.1 However, there are no clear guidelines on the choice of anticoagulant drugs, the period of administration, and even the need for anticoagulant therapy.7 In our case, we successfully treated our acute PVT patient with a 2-week subcutaneous injection of low molecular weight heparin and oral anticoagulant therapy.
In conclusion, our case shows that PVT can be provoked by protein C and S deficiency and that the PVT can be recanalized by short-term low molecular heparin plus oral warfarin therapy. Further study is needed to investigate the target INR, the period of administration, and the dosage and duration for relapse prevention in treatment with oral anticoagulants.

 XML Download
XML Download