

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Stroke is the major cause of chronic disability and the third leading cause of death in humans after cancer and coronary heart disease in developed countries. Because stroke causes severe neurological complications, new strategies for patients suffering from stroke are needed. Neuroprotection in ischemic stroke has been a topic of increasing concern and research. The capacity for neuroprotection depends on multiple factors including the use of an appropriate agent at the appropriate time and during the appropriate interval.1
A variety of animal models have been developed for modeling ischemic stroke. Among them, the most common model of focal ischemia in rats is the occlusion of the middle cerebral artery (MCAo) with an intraluminal suture, which mechanically occludes the major cerebral arteries.2 In contrast with other models of cerebral ischemia that involve mechanical occlusion of blood vessels, the rat photochemical cortical lesion models show endothelial damage, resulting in platelet aggregation, thrombosis, and permanent cerebral vascular occlusion.3 Considering that vascular thrombosis is responsible for more than 70% of human stroke cases and that only one Food and Drug Administration (FDA)-approved therapy exists for the treatment of acute ischemic stroke (the thrombolytic tissue plasminogen activator, or tPA), this model may be useful for understanding the pathobiology of ischemic stroke.4
Cerebral ischemia/reperfusion (I/R) injury triggers multiple and distinct but overlapping cell signaling pathways that may lead to cell survival or cell damage.5 Compared with global ischemia, focal ischemia is a reduction in blood flow to a very specific brain region as in embolic occlusion of the specific vessels.6 There are also significant differences in the modes of cell death between global and focal cerebral ischemia. In focal cerebral ischemia, most of the cells in the ischemic core undergo necrosis, but the rim of brain tissue that is hypoperfused surrounding the core is called the ischemic penumbra, which has the capacity to recover if perfusion is improved.7
In the penumbra, multiple mechanisms lead to apoptosis: excitotoxicity and ionic imbalance, oxidative stress, and apoptotic-like cell death may all occur and in both caspase-dependent and caspase-independent manners that may contribute to delayed ischemic cell death.8 A quite simplistic view implies poor prospects regarding cell survival in the core of the cerebral infarction and therapeutic expectations to control cell death and cell survival in the penumbra. In fact, focal cerebral ischemia triggers a cascade of molecular events that produce neuronal death ranging from immediate death to that occurring many days later, and this phenomenon raises the hypothesis that a "therapeutic window" exists during which interventions may improve neurological outcome.9 The aim of neuroprotection is to prevent delayed neuron apoptosis in the zone of the ischemic penumbra. On the basis of our current knowledge, many experimental trials have been reported to restore this process.10-12
In the photochemical stroke model, a ring lesion, which creates a cortical region of viable tissue surrounded by a circular rim of damaged tissue, has been described for the study of histological, biochemical, and molecular changes associated with the ischemic penumbra.13 TUNEL stain is used to demonstrate apoptotic and nonapoptotic features.14
Although the expression of pro- and anti-apoptotic factors and apoptotic cell death sequences have been extensively investigated, the time point changes in the mechanisms of cell death in photochemically induced focal cerebral ischemia remain inadequately defined. Previous investigations have reported on changes in the bcl-2 family in this model.14-16 Therefore, we examined changes in the expression of Bcl-2, Bax, caspase-3, phosphorylated Akt (pAkt), and survivin by Western blot 1, 3, and 7 days after photochemically induced focal cerebral ischemia in the rat.
MATERIALS AND METHODS
1. Animals and induction of focal cerebral ischemia
All surgical procedures and postoperative care were performed in accordance with the guidelines of the Chonnam National University Animal Care and Usage Committee. Twenty male Sprague-Dawley rats weighing between 200 and 250 g were used in this study. These animals were maintained on a 12-h light/dark cycle and were allowed free access to food and water. Each rat was anesthetized with 5% isoflurane and was maintained with 2% isoflurane in an oxygen/air mixture with the use of a gas anesthesia mask in a stereotaxic frame (Stoelting, Wood Dale, IL, USA). Focal cortical ischemia was induced by photothrombosis of the cortical microvessels using Rose Bengal (RB; Sigma Chemical Co., St. Louis, MO, USA) with cold light (Zeiss KL1500 LCD, Jena, Germany) as previously described with minor modifications.3,17 The expression of each apoptosis associated protein was measured 1, 3, and 7 days after the onset of ischemia. Body temperature was maintained during surgery at 37±0.5℃ with a heating pad controlled with a rectal probe. For illumination, a 4.5-mm fiber optic bundle from a cold light source was positioned on the exposed skull 0.5 mm anterior to the bregma and 3.7 mm lateral to the midline over the left sensorimotor cortex. The brain was illuminated for 10 min after infusion of 50 mg/kg of RB in normal saline into the right femoral vein via a microinjection pump within 1 min. The scalp was sutured and the rats were allowed to awake before being returned to their cages. The sham-operated control group received the same vein injection of RB and the same surgery, but without exposure to light; thus, no photochemical reaction was induced.3
2. Western blot for Bcl-2, Bax, Caspase-3, pAkt, and survivin
After decapitation, rat brains from different reperfusion time points (sham and days 1, 3, and 7) were quickly removed (each group, n=5). The ipsilateral and contralateral cerebral cortex was dissected. Samples were flash-frozen with liquid nitrogen and were stored at -80℃ until analyzed. Brain tissues were homogenized and proteins were purified by using 50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 60 mM n-Octyl-β-D-Glucopyranoside (OGP), and 1 mM phenylmethylsulfonyl fluoride (PMSF) supplemented with a protease inhibitor cocktail (Sigma). To prepare lysates, tissues were sonicated for 4 s for a total of five times and the homogenate was spun at 20,000×g for 20 min at 4℃ to remove insoluble material. Protein concentrations were determined with the BCA Protein Assay Reagent Kit (Pierce, USA) using bovine serum albumin (BSA) as the standard. Equal amounts of nuclear protein (20 µg) for Bcl-2, Bax, caspase-3, pAkt (serine-473), and survivin were separated on 6% to 12% SDS-polyacrylamide gels (USB Fueling Innovation, Cleveland, OH, USA). A PageRuler Plus Prestained Protein Ladder™ (Fermentas Life Sciences, Hanover, MD, USA) was used as a size reference. Proteins were transferred to pure nitrocellulose membranes (Bio-Rad, Richmond, CA, USA). The membranes were incubated in blocking buffer, 5% nonfat dry milk in TBST (200 mM Tris-HCl, pH 7.4, 500 mM NaCl, 0.2% Tween 20), at room temperature for 1 h with agitation. The membranes were then incubated with rabbit Bcl-2 polyclonal antibody (Santa Cruz, 1:2,000 in TBS with 0.2% Tween 20), rabbit Bax polyclonal antibody (Millipore, 1:3,000 in TBS with 0.2% Tween 20), mouse caspase-3 monoclonal antibody (Millipore, 1:1,000 in TBS with 0.2% Tween 20), polyclonal rabbit anti-phopho-Akt antibody (serine-472, Santa Cruz, 1:5,000 in TBS with 0.2% Tween 20), and mouse survivin monoclonal antibody (Santa Cruz, 1:1,000 in TBS with 0.2% Tween 20) overnight at 4℃. Following washes, the membranes were incubated with peroxidase-conjugated goat anti-mouse or anti-rabbit immunoglobulin (Santa Cruz Biotechnology, 1:8,000) in TBS with 0.2% Tween 20 for 1 h. After rinses in washing buffer, signal of bound antibodies was developed by an enhanced Immoblion Western chemiluminescent HRP substrate (Millipore, Bedford, MA, USA). Quantification of each band was performed by densitometry analysis (Scion, NIH software, Bethesda, MD, USA) of the protein signal using LAS 3000. Mouse anti-GAPDH monoclonal antibody conjugated with HRP (Sigma, 1:25,000) was detected on immunoblots as a loading control for protein quantitation.
3. Quantification and statistical analysis
To analyze the amount of the Bcl-2, Bax, caspase-3, pAkt, survivin, and GAPDH, we used a relative protein ratio to control, and quantitative data were expressed as means±SD. Differences between means were determined by one-way ANOVA followed by Dunnett's post hoc test for multiple comparisons. p<0.05 was considered statistically significant.
RESULTS
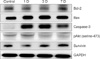
Ischemia caused changes in the expression of various apoptosis-associated proteins depending on the time of study (Fig. 1). Notably, the expression of Bax, caspase-3, and pAkt proteins was markedly increased in the ischemic hemisphere 1 day after the operation compared with that in the sham controls. The expression of Bcl-2 and survivin was slightly reduced at day 1 and then gradually increased by day 3. Bax expression, the bands at 21 kDa, reached peak levels at day 3 and gradually decreased by day 7. Caspase-3 expression, identified at 32 kDa, was abruptly increased from day 1 to day 3 compared with the sham control and decreased at day 7. pAkt was increased at day 1 but was slightly reduced from day 3. The Bcl-2 expression pattern was different at different time points. It was slightly reduced at day 1, but gradually increased from day 3, and was rising on day 7. However, the variation in expression of survivin, which is an inhibitor apoptosis protein (IAP), was insignificant compared with the others.
Changes in the density of each apoptosis-associated protein at each time point are shown in Fig. 2. Bcl-2 protein was increased 1.9-fold at day 3 (p<0.05 versus control) and increased 2.8-fold at day 7 (p<0.05 versus control) in the ischemic hemisphere. Bax and pAkt were increased 1.6-fold at day 3 and 1.8-fold at day 1, respectively (p<0.05 versus control). Caspase-3 was abruptly increased 8.4- to 9.9-fold from day 1 to day 3 (p<0.01 versus control), respectively, after the operation. No significant differences in survivin levels were found between sham controls and the experimental groups.
DISCUSSION
Focal and global cerebral ischemia in rats triggers apoptotic cell death within the ischemic lesion. In many previous reports,1,18 it was shown that new protein synthesis appears to be required for apoptosis. Comparison of human small cortical infarcts reveals identical histological patterns to photochemically induced rat brain lesions as well as naturally occurring human infarcts.19 Comparatively little is known about the apoptotic process in the photochemically induced model, so we evaluated the time point expression patterns of the major apoptosis regulatory proteins.
The Bcl-2 family of proto-oncogenes encode specific proteins such as Bcl-2, Bcl-XL, and Bax that regulate apoptosis. The anti-apoptotic effect of Bcl-2 occurs by prevention of cytochrome c release into the cytoplasm.5 Many studies have shown that over-expression of Bcl-2 can reduce ischemic brain injury in animal models of stroke.12,20 Activated Bax promotes cell death, unless it is bound by either Bcl-2 or Bcl-XL.21 However, the two molecules can act independently to exert their effects on cell death.5 In this study, we found strong induction of Bax protein 1 day after infarction in the ischemic hemisphere and it reached a peak on day 3. Compared with Bax expression, Bcl-2 protein expression was slightly reduced at day 1 and then abruptly increased at day 3 and was sustained at day 7. In a previous report using the same stroke model,15 Bax protein was in the cytoplasm of degenerating neurons in the center of the infarction between 4 h and 3 days after photothrombosis, and at the same time points, the levels of Bcl-2 and Bcl-XL proteins were markedly reduced in this region. These findings suggest that Bcl-2 and Bax protein expression switches around day 3 in this model. Interestingly, in the MCAo mouse model with preconditioning, Bcl-2 and Bcl-Xl were increased after sublethal forebrain ischemia but Bax remained unchanged.22 These differential expressions of the Bcl-2 family were associated with animal modeling, such as preparing of preconditioning, and reproducibility of lethal legion induction. The slight decrease in Bcl-2 expression within 1 day might also be attributed to postischemic protease activation.23
Caspases are cysteine proteases that are constitutively expressed as zymogens or pro-caspases and that are specifically activated during apoptotic stimuli.24 Several previous studies reported that the role of caspase-3 is as a key mediator of apoptosis in ischemic stroke.25 Briefly, upregulation of caspase-3 mRNA in rat brain occurs 1 h after the onset of focal ischemia, whereas caspase-3 and its cleavage products in mouse brain are activated during early reperfusion 2 h after MCAo. Ischemic damage could be reduced by caspase inhibitor treatment even 9 h after MCAo. We observed a significant increase in the cleaved active form of caspase-3 from day 1 of operation until day 3, when the maximal intensity was observed (Fig. 2C). Also, this increased caspase-3 expression was sustained at day 7. We did not observe any significant change in the contralateral hemisphere. These findings are in agreement with an extended treatment window for caspase inhibition after stroke.18
Akt is a subfamily of serine/threonine protein kinases and is activated via phosphatidylinositol-3 kinase (PI3K). After phosphorylation, Akt promotes cell survival and prevents apoptosis by inactivating several targets. In the MCAo model,26 phosphorylation of Akt (pAkt) was temporally accelerated at serine-473 in the ischemic cortex after transient focal cerebral ischemia and was then decreased by 24 h. However, in the ischemic core, pAkt was decreased only after ischemia. pAkt-positive cells were colocalized with NeuN-positive cells in the cortex, but pAkt-positive cells did not co-localize with TUNEL-positive cells. In this study, pAkt expression was increased about 2-fold at day 1 and was decreased by day 3 compared with the sham control. This change in pAkt expression could be explained by the propagation of the ischemic lesion. The infarct develops in the superficial cortex, whereas the higher-order branches of the larger patent vessels in the deeper cortical layers are occluded by tissue compression caused by the vasogenic edema associated with this procedure.27 Further, complete wedge-shaped coagulative necrosis was formed 24 h after ischemia according to histopathological examination.28 Thus, as in previously reported studies,26 pAkt expression in the photochemical-induced stroke model might also depend on the severity of the ischemia.
Survivin is a member of the IAP family that serves multiple functions related to both cell survival and cell division within many cell types and only recently has come under study in relation to ischemia in heart or brain.29 Recently, Zhang et al.30 reported that this protein is a target for the PI3-kinase/Akt pathway and can antagonize the effects of apoptosis-inducing factor. On the basis of current knowledge, we hypothesized that survivin would be significantly changed in our stroke model, but as noted, the changes in survivin expression evaluated by Western blot were insignificant. In fact, infarct volumes in survivin+/-. mice at 24 h and 3 d were not significantly different from those of survivin+/+ mice, which suggests that the role of survivin in neuroprotection is minimal.29
In this study, we evaluated the expression of apoptosis regulatory proteins in photochemically induced focal cerebral ischemic rat brain. To interpret these changes, we have to consider the changes in cerebral blood flow (CBF) after photothrombosis. In rats subjected to a photothrombotic stroke lesion, cortical CBF at the center of the region at risk was gradually reduced until 48 h after irradiation with flow values of 33%. However, a significant partial restoration of blood flow compared with cCBF values at 24 h was observed at 72 and 96 h after irradiation. Hence, cortical CBF was 56% of preischemic levels at 72 h and 87% at 96 h after ischemic induction. These CBF changes could explain the meaningful changes in Bcl-2, Bax, and caspase-3 around day 3.
In conclusion, targeting and preventing apoptosis in the penumbra seems to be a rational therapeutic goal for reducing cerebral infarct volume after clinical stroke, and the present study may provide the basis for the design of therapeutic interventions using the photochemical thrombosis stroke model. However, the expression of each apoptosis regulatory protein as well as the regional distribution and association with DNA fragmentation should be assessed further.

 XML Download
XML Download