

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Farber disease (FD), named after Sidney Farber in 1957, is a lysosomal storage disorder caused by a deficit of acid ceramidase that is encoded by the ASAH1 gene [1]. It shows autosomal recessive inheritance, and its prevalence has been estimated to be lower than 1/1,000,000 [2]. Most patients with this disease show unique symptoms, such as painful deformed joints, subcutaneous nodules, and progressive hoarseness, in early infancy, and progression of this disease often leads to death within a few years after birth [3].
The treatments of FD are mainly focused on relieving symptoms and pain with medication or surgical correction. Although hematopoietic stem cell transplantation (HSCT) has been suggested to prevent progression of FD [4,5], there is no effect on symptoms related to the central nervous system, which are considered the most critical symptoms in patients with classic FD. Therefore, HSCT is limited to FD type 2 and 3 that do not involve the central nervous system, and treatment reports are limited [5].
Here, we report the case of a female patient with FD, who was misdiagnosed with epithelioid hemangioendothelioma (EHE) and was treated for EHE until four years of age.
CASE REPORT
The patient was a 4-year-old (58 months) girl. She was born at 37 weeks of gestation, and her birth weight was 3,400 g. She was transferred to the neonatal intensive care unit (NICU) immediately after birth because she had seizure-like movements and respiratory difficulty. Initially, floppy infant syndrome was diagnosed, as she showed hypotonia signs, such as frog position and weakness of the extremities. Echocardiography showed congenital heart diseases (atrial septal defect, ventricular septal defect, and patent ductus arteriosus); therefore, she underwent cardiac surgery 40 days after birth. As her respiratory and cardiac status improved, she was discharged two months later, although hypotonia remained.
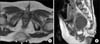
She visited a pediatric neurosurgery outpatient clinic at 11 months after birth because of multiple systemic granulomas and a sacrococcygeal mass. T1-weighted magnetic resonance imaging showed a subcutaneous enhancing lesion (3.0×2.0×3.4 cm) at the sacrococcygeal level (Fig. 1). She was transferred to our department with a suspicion of sacrococcygeal teratoma. She underwent surgery, and the mass was completely resected at 13 months of age. On pathological examination, we diagnosed the mass as malignant EHE. There were regions with diffusely infiltrating epithelioid cells, and we noted epithelioid markers, such as CD31 and Fli-1, in the mass. Additionally, nuclear pleomorphism and mitosis were noted, with a high Ki-67 index, which suggested malignancy.
After the first operation of the sacrococcygeal mass, she underwent redo-excision at 17, 21, and 28 months after birth for recurrent sacrococcygeal masses. In addition, she also underwent scalp mass excision at 21 months of age (2 lesions, right and left occipital regions), 27 months of age (right parieto-occipital region), and 33 months of age (left occipital region). At these time points, we considered the scalp masses as metastatic EHE in pathologic reviews.
From 13 months of age, the patient underwent repeated mass excisions in the sacrococcygeal and scalp areas, and we performed medical management of EHE during this period. Interferon-α was used between 18 and 21 months of age, and oral cyclophosphamide was administered between 42 and 47 months of age to suppress recurrently growing lesions [6,7]. However, these therapies did not show any significant effects.
As she became older, she showed difficulty in food ingestion and speaking. Additionally, joint deformities and stiffness were noted in most of her joints. Subcutaneous granulomas were generalized, and they were observed at the scalp, chin, digital joints, sacral region, back, and feet (Fig. 2). We suspected other diseases, and therefore, we performed whole genome exome sequencing at the age of 49 months, and an ASAH1 gene mutation, a characteristic finding of FD, was identified.
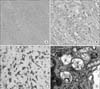
When the patient was 50 months of age, a recurred sacrococcygeal mass was excised again. On H&E staining of the specimen obtained from the mass, dense sclerotic collagenous tissues infiltrated with epithelioid cells (Fig. 3A, B) were noted, and endothelial cell markers, such as CD31 and Fli-1, were noted on immunohistochemical (IHC) analysis. There were no findings indicative of malignancy, such as severe atypism and a high level of mitosis. In addition, on IHC analysis, strong cytoplasmic staining of CD68 was noted, which indicated the presence of macrophages rather than epithelioid cells (Fig. 3C). On transmission electron microscopy, there were numerous spindle-shaped cells with enlarged lysosomes and C-shaped curvilinear tubular bodies in a collagenous background (Fig. 3D), which are also characteristic findings of FD.
Presently, although she is 58 months old, her developmental status reflects an age of only 12 months. On radiological examination, we identified definite brain parenchymal atrophy. Her height and body weight are below the third percentile of children aged four years. Joint stiffness was noted at both elbows (range of motion [ROM], 160°), both knees (ROM, 100°), the hip joint (ROM, 120°), and both sides of the third and fourth interphalangeal joints.
DISCUSSION
Although FD is a genetic disease, there was no family history of FD in our patient. A family history is generally not noted because FD is expressed when mutated genes are inherited from both parents, as this is an autosomal recessive disorder. Additionally, there is a possibility of spontaneous mutation.
The most common subtype of FD is type 1, and patients with type 1 FD show various neurological disorders together with the three major symptoms of painful joints accompanied by deformities and stiffness, multiple subcutaneous granulomas, and vocal changes from an early age (usually two weeks to four months after birth) [8]. Our patient showed neurological disorders, such as limb weakness, mental retardation, and definite brain parenchymal atrophy, in addition to the symptom triad indicative of classical FD. Therefore, we believe that our patient had type 1 FD.
In FD patients, subcutaneous granulomas might occur at regions that experience mechanical pressure, and joints, such as the interphalangeal joint, wrist, elbow, and ankle, are common sites [9]. Our patient had granulomas at the scalp, chin, ears, interphalangeal joints, feet, and sacrococcygeal joint. She underwent five surgeries for sacrococcygeal masses and three surgeries for scalp masses.
Voice change or hoarseness occurs mainly due to protrusion of a granuloma at the larynx, which results in compression of the vocal cords as the granuloma grows. Because most FD patients die from aspiration pneumonia or dyspnea caused by lesion invasion in respiratory organs, identification of lesions in the respiratory tract is important. If a patient is suspected with pneumonia, prophylactic management of pneumonia would be very important to prevent an adverse prognosis. It has been reported that approximately 25% of FD patients have hepatomegaly or splenomegaly [9].
There is no specific treatment for FD patients. However, HSCT, which improves ceramidase production, is emerging as a new treatment choice for FD. Unfortunately, it can be considered for only type 2 or 3 FD patients, who have no neuronal degeneration or a lower degree of neuronal degeneration than that in type 1 FD, because HSCT does not improve neuronal degeneration [5]. Therefore, in our patient, we expected HSCT to have limited effect because central nervous system symptoms and developmental delay were present.
Although most FD patients die within two years after birth, some patients have been reported to live much longer than expected [10]. Our patient reached 58 months of age, and in such patients, modalities for effective management of the disease should be considered to ensure longer survival.
Our patient was misdiagnosed with malignant EHE initially, and unnecessary medication was administered and excessive surgery was performed. Based on this case, we believe that if a patient has recurrent and atypical findings, which cannot be explained by the current diagnosis, another disease should be strongly suspected, even if the final diagnosis has been made. Accurate diagnosis of the patient’s condition would help in the treatment of pain and the appropriate management of the disease, although this involves additional effort and expenses.
In conclusion, we reported the case of a female patient with FD, who was misdiagnosed with EHE and was treated for EHE until four years of age.

 XML Download
XML Download