

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pancreatic tumors constitute less than 1% of all neoplasms and have a poor prognosis. Neuroendocrine and acinar cell carcinomas account for approximately 7% and 1% of pancreatic cancers, respectively, whereas ductal adenocarcinomas account for more than 75%. Among the many pathological subtypes of pancreatic tumors, there are less than 20 reported cases of the mixed endocrine-exocrine type in the English literature [1]. Pathologically, such tumors are termed mixed acinar-neuroendocrine carcinoma (MANEC), in which neuroendocrine cells comprise more than 30% of the tumor mass. Although there are no definite therapeutic guidelines for MANEC, complete surgical resection is widely suggested to be the only way to improve survival in patients with pancreatic malignancies [2]. In this study, we report a 13-year-old boy with MANEC who initially presented with periumbilical pain.
CASE REPORT
A 13-year-old boy presented to our emergency room with persistent vomiting that began 5 days previously along with poor oral intake and periumbilical squeezing pain. The vomiting persisted regardless of whether he ate. The boy was able to pass gas and defecate normally. He had a palpable mass on his abdomen with obscure tenderness and hypoactive bowel sounds. He did not have any family or past operational histories, except that the patient had multiple enchondromatosis. His WBC count (10,020/µL) and CRP (5.96 mg/L) were elevated slightly above the normal range, whereas tumor marker levels of CA 19-9, CEA, and β-human chorionic gonadotropin were not elevated. Human chorionic gonadotropin, amylase, and lipase levels were within the normal range, which suggested that the mass was a non-functioning tumor.
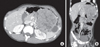
A 9.8×10.0-cm mass of uncertain origin was detected close to the pancreas and spleen on abdominal ultrasonography and abdominal-pelvic CT. The mass was lobulated and heterogeneously enhanced with a multifocal cystic portion around the spleen (Fig. 1). Fine needle aspiration biopsy (FNAB) was planned and conducted to determine the possibility of neuroendocrine tumor and pancreatoblastoma. However, we could not make a definitive diagnosis; pancreatoblastoma was suspected based on the presence of beta-catenin positive cells. There were no remarkable findings on a preoperative chest CT or a bone scan to detect metastasis.
Abdominal cavity exploration revealed that the mass extended from the body and tail of the pancreas to the transverse colon and splenic hilum. Hence, distal pancreatectomy, segmental resection of the transverse colon, and splenectomy were performed for complete curative resection.
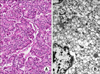
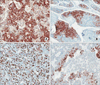
The patient started a standard diet 9 days postoperatively and was discharged the following day. Pathological diagnosis of the tumor determined a mitotic count of 50 cells/10 HPF; 30% of the tissue was necrotic, and the tumor cells were positive for chromogranin A and synaptophysin, which supported the final diagnosis. Ki-67 proliferative staining was positive in 80% of cells, and the tumor cells were positive for beta-catenin, CD10, E-cadherin, and cytokeratin (Fig. 2, 3). The final pathological diagnosis was mixed acinar-endocrine carcinoma. The patient is currently undergoing adjuvant chemotherapy with cisplatin, etoposide, and cyclophosphamide in our pediatrics department; the regimen of chemotherapeutic agents used was based on the standard regimen used to treat pancreatoblastoma.
DISCUSSION
Mixed endocrine-exocrine tumors of the pancreas are rare. Of the 20 cases reported in the literature, most were reported in adults aged more than 60 years [1]. Most pancreatic malignancies in children are pancreatoblastomas; only a single case of MANEC has been previously reported, in an 8-year-old boy [3]. MANEC is more common in women, although the number of reported cases is small [456]. Most patients present with abdominal pain and weight loss [378]. A standardized management protocol has not been established owing to the small number of cases, but complete or debulking surgical resection is generally considered the first line treatment, after which additional chemotherapeutic agents can be applied [3]. The prognosis of patients with MANEC is generally poor, with a mean survival of 10.5 months after surgical resection of the primary tumor [3].
Pancreatic carcinomas with an acinar cell phenotype such as acinar cell carcinomas, pancreatoblastomas, and mixed carcinomas represent less than 2% of adult pancreatic neoplasms [2]. MANEC is usually diagnosed when the proportion of neuroendocrine cells within the tumor exceeds 25%, which is determined by morphology and/or immunohistochemical staining for chromogranin A or synaptophysin. Several studies have compared the characteristics of MANEC with those of other malignant pancreatic tumors. MANEC has a wide range of pathological characteristics [910]. For example, some mixed acinar-ductal carcinomas exhibit intracellular or stromal mucin as evidence of ductal differentiation, whereas others have separate components of individual glands surrounded by desmoplastic stroma [10]. However, the majority of reported cases have shown a predominantly acinar pattern.
This case required surgical biopsy to make a definitive tumor classification, as fine needle biopsy was insufficient. Because of the small number of MANEC cases and immunophenotypes that overlap with other diseases, accurate diagnosis of rare pancreatic tumors by cytology can be challenging. Previous studies reported the sensitivity and accuracy of endoscopic ultrasonography (EUS)-guided fine needle aspiration (FNA) in detecting malignancy as 75%-92% and 79%-92%, respectively. However, EUS-guided FNA has been associated with some drawbacks, and a systemic review of 53 studies estimated a negative predictive value of 60%-70% for pancreatic adenocarcinoma [11]. Treatment protocols are lacking, not only for MANEC, but also for acinar cell malignancies [12]. In one study regarding mixed acinar endocrine ductal carcinoma, 10 of 11 patients underwent primary surgery including pancreatoduodenectomy, distal pancreatectomy, and splenectomy. Of those patients, some underwent adjuvant therapy, and most were administered chemotherapy [2]. In the present case, the patient is undergoing a regimen of postsurgical adjuvant chemotherapy based on the standard regimen for pancreatoblastoma treatment.
We reviewed 7 reports in the PubMed database published between 2007 and 2014. Of these, 17 cases had pancreatic masses classified as MANEC or mixed acinar ductal carcinoma [12312131415]. In those reports, which included 5 female and 12 male patients, the mean patient age was 65.6 years and the mean mass size was 4.0 cm, with an unreported mass size in 1 case. The majority of tumors were in the head of the pancreas, 2 were in the tail, and 1 was in the body. In the present case, the tumor spanned the body and tail of the pancreas. Fifteen patients underwent surgery including pancreatectomy and splenectomy. With a mean follow-up of 24.2 months, the outcome varied from alive without disease to dead due to disease.
In summary, we report a rare case of MANEC in a 13-year-old boy who presented with a palpable mass. FNAB was not useful for making a definitive diagnosis, and MANEC was confirmed pathologically after complete surgical resection. To provide a clearer therapeutic protocol for MANEC, more cases need to be reported.
 XML Download
XML Download