

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Lung cancer is a common cancer and life-threatening disease in Korea. The mortality of lung cancer is highest than colon, breast, or prostate cancers [1234]. Lung cancer is classified as two types which are small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). The molecular pathogenesis of SCLC and NSCLC is different. SCLC accounts for 15% of lung cancer cases and it is a highly malignant and clinically aggressive tumor. On the other hand, 85% of lung cancers are diagnosed as NSCLC and has three major subtypes: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma [3567].
A lot of agents have been used for clinical treatment of lung cancer [7]. One of the targets is the cyclooxygenase (COX) which is evaluated in the treatment of lung cancer. COX is essential enzyme for the prostaglandins (PGs) synthesis. PGs are involved in various physiological and pathogenetic processes such as inflammatory reaction, gastro-intestinal cytoprotection, ulceration, hemostasis, and thrombosis [891011121314]. COX enzyme has two isoforms, COX-1 and COX-2. While COX-1 is consistutively expressed in most cells and tissues, COX-2 is expressed in central nervous system, kidney, and seminal vesicles. However, it can be induced by various proinflammatory and mitogenic stimuli [8910].
To regulate function of COX, non-steroidal anti-inflammatory drugs (NSAIDs) have been used traditionally for several decades. NSAIDs are agents for treatment of pain, inflammation, and fever [1215]. NSAIDs has been used to manage pain and suppress inflammation in arthritis, but these days they are also used as the agents for cancer therapeutics. NSAIDs have contributed to anti-proliferation and pro-apoptosis on a various cancer cells, such as colorectal cancer, breast cancer, or gastric cancer [8111213161718]. NSAIDs block a production of PGs by the inhibition of COX-1 and COX-2 in normal and inflammatory tissues. There are side effects such as gastrointestinal tract bleeding and kidney failure when NSAIDs are treated for a long time [12151819]. These side effects are known by inhibition of COX-1 [1219]. On the other hand, COX-2 inhibitors are widely used because they have little side effect. There are several selective COX-2 inhibitors, for example rofecoxib, etoricoxib, and celecoxib. The celecoxib makes little effects for COX-1, and inhibits tumor initiation and tumor cell growth. In past studies, celecoxib was reported which decreases risk of colon, breast, esophagus, and gastric cancer, it is also effective for chemotherapy and radiotherapy of cancer patients [16]. Celecoxib induces cell cycle arrest, inhibits tumor growth, suppresses tumor angiogenesis and induces apoptosis in several types of cancers [141618]. Moreover, celecoxib induces apoptotic cell death in tumor cells and endothelial cells [111619]. Therefore, investigation of mechanism on celecoxib-induced apoptosis is important to understand anti-cancer activity.
Recently, endoplasmic reticulum (ER) stress has been reported as a major mechanism that celecoxib can induce cell death in cancer [2021222324]. Unfolded protein response (UPR) is a pro-survival mechanism and remains homeostasis of cells in ER stress state. However, if this adaptation is not proven to be sufficient, the apoptosis starts [20212526]. Several solid tumors undergo ER stress and are going to fall a fate, such as adaptation or apoptosis. Three types of ER transmembrane proteins are important: protein-kinase and site-specific endoribonuclease inositol-requiring enzyme 1 (IRE1), protein-kinase R-like ER kinase (PERK)/pancreatic eIF2 kinase and activating transcription factor 6 (ATF6) [202126]. C/EBP homologous transcription factor (CHOP) is a major transcription factor of pro-apoptosis and regulates ER stress-induced apoptosis. CHOP induces cell death through controlling the expression of other genes [2021]. ER stress is induced by NSAIDs treatment which causes ER membrane protein PERK to phosphorylate eIF2α. CHOP is up-regulated after the phospho-eIF2α protein activates ATF4. NSAIDs also induces apoptosis through another complicated mechanism as well as ER stress [19].
Taken together, celecoxib affects obviously cell death of lung cancer but its mechanism is unclear. In this study, the involvement of ER stress in cell death is investigated when celecoxib is treated on lung cancer cells.
Materials and Methods
Cell culture
Human lung carcinoma cell line A549 and H460 (ATCC, Manassas, VA, USA) were maintained in RPMI1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Hyclone) and 100 U/ml penicillin, and 100 µg/ml streptomycin (Hyclone) at 37℃ in humiditied 5% CO2 atmosphere.
Analysis of cytotoxicity
Cell viability was examined through the WST-1 assay (Takara, Shiga, Japan). Cells were seeded in 96-well plates and incubated for 24 hours. Celecoxib (TRC, Toronto, Ontario, Canada) of various concentrations was treated on cells for overnight. WST-1 was added each well and the plates were incubated for 1 hour and the absorbance was measured using microplate reader (Thermo Pierce, Waltham, MA, USA) at 450 nm wavelength.
Annexin V assay
Cells were collected by centrifugation and washed with phosphate buffered saline (PBS). Fluorescein isothiocyanate (FITC)-labeled annexin V (BD Bioscience, San Jose, CA, USA) was added in dark after cell pellet was resuspended with annexin V binding buffer. Cells were detected by flow cytometry (Miltenyi Biotech, Bergisch Gladhach, Germany) after incubation for 15 minutes. To assess involvement of caspase cascade, Z-VAD-fmk (Merck, Darmstadt, Germany) was treated for 1 hour before celecoxib treatment.
RNA interference
A549 cells were transfected with glucose-regulated protein 78 (GRP78), IRE1, or negative control siRNA (GenePharma, Shanghai, China) according to manufacturer's instruction. A549 cells were seeded in 6-well plates with growth medium without antibiotics and allowed to reach 50% confluence for transfection. Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and siRNA were respectively diluted in medium without serum and incubated for 5 minutes at room temperature. Two solutions were mixed and incubated for 20 minutes at room temperature. The mixture was treated on cells for 16 hours.
Reverse transcription polymerase chain reaction
Total RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's protocol. Briefly, harvested cells were mixed TRIzol and added chloroform. The mixture was centrifugated at 13,000 rpm for 15 minutes at 4℃. Upper solution of the separated two layer was transferred new tube and combined with isopropanol. After incubation for 10 minutes, combined was centrifugated for 15 minutes at 4℃. Supernatant was discarded and washed with 75% ethanol in diethylpyrocarbonate (DEPC) treated water. RNA containing pellet was resuspended with DEPC treated water. RT-premix (Bioneer, Daejeon, Korea) was used for cDNA synthesis from total RNA. cDNA was amplified using primers and Tenuto PCR premix (Enzynomics, Daejeon, Korea) in PCR Thermal Cycler (Takara). PCR products were analyzed by gel electrophoresis in TAE buffer through agarose gel.
Western blot analysis
Cells were lysed in lysis buffer (Invitrogen) supplemented with protease inhibitor cocktail (Sigma, St. Louis, MO, USA) after the cells were harvested. Samples were incubated on ice for 10 minutes and centrifugated for 10 minutes at 13,000 rpm. Proteins were determined by the bicinchoninic acid assay (Thermo Pierce) and were separated in 10% sodium dodecyl sulfate gel overlaid with a 4% polyacrylamide staking gel. Proteins were transferred to a nitrocellulose membrane after separation. The membrane was blocked with 5% skim milk for 1 hour at room temperature and washed with Tris-buffered saline containing 0.1% Tween 20 (TBST, pH 7.4) for 10 minutes each time for three times. The membrane was incubated with primary antibodies of caspase 3 (Cell Signaling, Beverly, MA, USA), caspase-4 (Abcam, Cambridge, MA, USA), caspase 8 (Cell Signaling), caspase 9 (Cell Signaling), GRP78 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), CHOP (Cell Signaling), and β-actin (Santa Cruz Biotechnology) in 0.1% TBST at 4℃ overnight. The membrane was incubated with secondary antibody (anti-goat IgG for GRP78; anti-rabbit IgG for caspase 3 and caspase 9; anti-mouse IgG for caspase 8, CHOP and β-actin at 1:5,000 dilutions) for 1 hour at room temperature after the membrane was washed with 0.1% TBST three times for 10 minutes. Following incubation, membrane was washed with 0.1% TBST three times. The band signals were detected by chemiluminescence (Fuji Photo Film Co., Tokyo, Japan).
Results
Effect of celecoxib on cell death of lung cancer cells
Cytotoxicity was examined when celecoxib was treated on lung cancer cells. A549, H460 lung cancer cell lines were incubated with various concentration of celecoxib (0, 25, 50, 100, and 200 µM). Cell viability was observed by WST-1 assay after overnight treatment of celecoxib.
Approximately, the half maximal inhibitory concentration (IC50) of celecoxib was between 50 and 100 µM (Fig. 1). To determine whether celecoxib induces apoptosis, celecoxib (50 and 100 µM) was treated on A549 and H460 cells for overnight and then analyzed by flow cytometry after staining with FITC-labeled annexin V. Apoptosis was induced on A549 and H460 cells which were treated 100 µM of celecoxib. The rate of apoptosis by celecoxib was 61.7% on A549 and 25.5% on H460 respectively when 100 µM of celecoxib was treated (Fig. 2).
Involvement of caspases in celecoxib-induced apoptosis
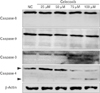
Celecoxib-induced apoptosis on lung cancer cells (Fig. 2). To investigate that celecoxib induces apoptosis through caspases, celecoxib was treated on A549 cells. It was observed whether pan-caspase inhibitor, z-VAD-fmk reduced celecoxib-induced apoptosis. The z-VAD-fmk was treated for 1 hour before treatment of celecoxib. Celecoxib-induced apoptosis was decreased at pretreatment with z-VAD-fmk than only treatment with celecoxib (Fig. 3). Then, caspases were detected using western blot after celecoxib treatment on A549. Caspase-3, -8, and -9 did not be activated, but caspase-4 was cleaved with treatment of 75 and 100 µM celecoxib (Fig. 4).
Involvement of ER stress in celecoxib-induced apoptosis
To examine whether celecoxib-induced apoptosis is involved in ER stress, markers of ER stress were investigated. These markers include GRP78/immunoglobulin heavy chain binding protein (BiP), CHOP/growth arrest and DNA damage inducible protein (GADD) 153, ATF4, GADD34, and spliced X-box binding protiens-1 (sXbp-1). According to the results of reverse transcription polymerase chain reaction, expression of GADD34 and CHOP was increased but GRP78, sXbp-1, and ATF4 was not in A549 cells. GRP78, sXBP-1, GADD34, and CHOP was increased in H460 but ATF4 was not increased (Fig. 5A). Also, GRP78 and CHOP was increased in protein level in A549 (Fig. 5B).
UPR induces transcription of folding chaperones. However, ER stress signaling pathways trigger cell apoptosis when UPR is continued. These signaling pathway-related molecules are IRE1, PERK, and ATF6. GRP78 regulates pro-survival pathway in UPR and is important to protein folding and assembly. To detect whether GRP78 is related to celecoxib-induced apoptosis, GRP78 was inhibited using by siRNA. Proportion of apoptotic cells induced by 75 µM of celecoxib was 25.86% and apoptosis was increased (42.54%) when GRP78 siRNA and celecoxib were combined (Fig. 6). When sXBP-1 as a downstream effector of IRE1α was inhibited by siRNA, the rate of apoptosis was not changed (27.55%).
PERK induces ATF4 and CHOP after eIF2α is phosphorylated. To examine whether PERK pathway was played by celecoxib, salubrinal, inhibitor of eIF2, was pre-incubated for 1 hour and Annexin V was detected by flow cytometry after 100 µM of celecoxib treatment. The rate of apoptosis was little changed on celecoxib treatment A549 cells when eIF2 was inhibited (Fig. 7).
Discussion
NSAIDs are most common drugs that reduce pain and inhibit synthesis of PGs. NSAIDs have been contributed anti-proliferation and pro-apoptosis on a various cancer cells. This study found out how lung cancer cells undergo celecoxib-induced apoptosis. It was confirmed that ER stress is a key role in celecoxib-induced apoptosis in lung cancer cells.
Evidences that NSAIDs induce cell death are reported in many studies. Other studies observe that aspirin and indomethacin induce apoptosis on gastric cancer [27], aspirin induces apoptosis on ovarian cancer [28], and NS398 induces apoptosis on colon cancer [29]. As shown in Fig. 1, celecoxib among NSAIDs induced cell death on H460 by dose-dependent manner. However, when celecoxib was treated on A549 cells, cell viability was slightly increased at low concentration of celecoxib (25 µM). In other study, cell death was observed when 30 µM of celecoxib was treated on A549 cells [30]. Although WST-1 assays for celecoxib was retried on A549 cells several time, similar results that viability was increased at low concentration of celecoxib were detected. The low concentration of celecoxib might regulate other signal pathways and then temporarily increase cell viability. To find solution to this problem, other investigations are needed.
This study focused the mechanism of celecoxib-induced apoptosis on lung cancer. Several studies suggest that NSAIDs-induced cell death is related with ER stress in various cancer cells [222631323334]. UPR induces transcription of genes encoding ER resident chaperones. However, if ER stress is continuously persisted, signaling pathways including PERK, IRE1, and ATF6 trigger apoptosis. In this study, two of three pathways involved in ER stress-induced apoptosis were determined. Celecoxib-induced apoptosis might be less related to PERK pathway because salubrinal, eIF2α inhibitor did not block apoptosis (Fig. 7). The rate of apoptosis was also not changed by inhibition of IRE1 on lung cancer cells (Fig. 6). siRNA for CHOP also did not diminish celecoxib-induced apoptosis (data not shown), although CHOP as an important effector of ER stress was increased after celecoxib treatment. In contrast, inhibition of GRP78 increased apoptosis of lung cancer cells. Continuous ER stress consumes GRP78 and subsequently increases GRP78 production; thus, down-regulation of GRP78 by siRNA might aggravate ER stress conditions. This result suggests that celecoxib-induced apoptosis is deeply associated with ER stress depending on GRP78.
However, other studies suggest that celecoxib-induced apoptosis is involved in caspase cascade [171834]. Liu et al. [34] investigated that celecoxib treatment appears to activate the caspase cascade in lung cancer cells. They showed that apoptosis with celecoxib was involved in cleavage of upstream and downstream of caspase cascades, caspase-3, -8, -9, and poly(ADP-ribose) polymerase, so celecoxib is regarded to induce apoptosis through ER stress and caspases cascade. Actually, we investigated that z-VAD-fmk, pan-caspase inhibitor, reduced celecoxib-induced apoptosis but celecoxib did not activate caspase-3, -8, and -9 on lung cancer cells (Fig. 4). ER stress is also associated with non-classical caspases such as caspase-4 [35]. We found that celecoxib activated caspase-4 on A549 (Fig. 4).
Taken together, celecoxib triggered ER stress on lung cancer cells and celecoxib-induced apoptosis might be involved in both non-classical caspase-4 and GRP78. These finding could provide an insight of a therapeutic approach using celecoxib as an adjuvant with anti-cancer drugs.
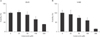

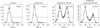



 XML Download
XML Download