

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cisplatin is one of the most effective chemotherapeutic agents for the treatment of various malignant tumors [1]. Although the mechanism of its anticancer activity is not fully elucidated, it binds to DNA and then forms DNA inter-/intra-strand cross-links in cancer cells, resulting in an arrest of DNA synthesis and replication [2]. The cross-link can induce DNA damage, if the damage is extensive, and lead to cell death. Because of this, the use of high-dose cisplatin in cancer therapy is limited due to its severe side effects in normal tissues, such as kidney, nerve, inner ear, and red blood cells. Among them, nephrotoxicity is a major concern. After cisplatin treatment, approximately one-third of cancer patients develop renal dysfunction, as represented by lower glomerular filtration rate and higher serum creatinine concentration, resulting in acute renal failure [3]. It has been demonstrated that the cytotoxic effect of cisplatin occurs via several mechanisms, including DNA damage, mitochondrial injury, and transcriptional dysfunction in kidney tubular cells, as well as tumor cells [4]. The etiology of cisplatin cytotoxicity is associated with oxidative stress. Many studies have shown that cisplatin causes oxidative stress in kidney tubule epithelial cells [45], but the molecular mechanism is not fully understood. Therefore, a deep understanding of the redox system during nephrotoxicity induced by cisplatin is of critical importance for the improvement of cancer chemotherapy.
Poly(ADP-ribose) polymerase (PARP) catalyzes a reaction in which the ADP-ribose moiety of NAD+ is transferred to a receptor amino acid, forming poly(ADP-ribose) polymers. A family of human PARP is divided into 17 enzymes sharing a conserved catalytic domain [6]. Among them, PARP1 is responsible for the majority (approximately 90%) of PARP activity. The ubiquitous and abundant nuclear enzyme PARP1 plays multiple roles in the regulation of chromatin structure, transcription, protein function, and protein-protein interaction [6]. While historically PARP1 has mainly been described as the key enzyme for DNA damage detection and repair, its excessive activation pathologically leads to necrotic cell death through the depletion of intracellular ATP [7]. Genetic or pharmacological inhibition of PARP1 protects against various kidney diseases, including ischemia and reperfusion injury [8], ureteral obstruction [9], tubulointerstitial fibrosis [10], diabetes [11], and glomerulonephritis [12]. In terms of cisplatin nephrotoxicity, our previous reports have demonstrated that loss of PARP1 is not only protective against kidney structural/functional damage and inflammation through blocking of the toll-like receptor 4/p38/tumor necrosis factor-α axis [13], but also treatment with PJ34, a potent PARP1 inhibitor, by respective preconditioning and postconditioning reduces such kidney injuries [131415]. Furthermore, loss of PARP1 reduces oxidative stress assessed by the concentration of lipid hydroperoxide (LPO) and the ratio of reduced glutathione to oxidized glutathione during cisplatin nephrotoxicity [13]. It has been generally postulated that the generation of oxidative stress triggers excessive PARP1 activation through DNA damage [16]. Conversely, it has remained unclear by what mechanism PARP1 inhibition reduces oxidative stress.
Sirtuin (SIRT) is a family of NAD+-dependent enzymes that remove acetyl groups from lysine residues on specific protein substrates. In an antioxidant defense system, SIRT3 regulates acetylation and activity of key enzymes including manganese superoxide dismutase (MnSOD) [17], catalase [18], glutathione peroxidase (GPX) [19], and isocitrate dehydrogenase 2 [20]. Many studies have demonstrated that SIRT3 is a key regulator of cell defense and survival in response to oxidative stress induced by various injuries [212223]. In a recent report using a mouse model of cisplatin nephrotoxicity, SIRT3-deficiency has shown to increase during kidney dysfunction and mortality [24]. Intriguingly, it has been reported that SIRT3 downregulation involves PARP1 activation in hyperglycemia-injured endothelial cells [25]. Therefore, we hypothesized that PARP1 inhibition improves antioxidant status through SIRT3 activation during cisplatin nephrotoxicity. To investigate this hypothesis, we sought to determine whether PARP1 inhibition could alter SIRT3 expression and activity, and if so, whether loss of SIRT3 could erase the protective effect of PARP1 in cisplatin-injured kidney tubular cells.
Materials and Methods
Cell culture
Human kidney 2 (HK2), a proximal tubular cell line derived from normal kidneys, was obtained from the American Type Culture Collection (Rockville, MD, USA) and maintained in keratinocyte-serum-free medium containing 5 ng/ml human recombinant epidermal growth factor and 40 µg/ml bovine pituitary extract (Life Technologies, Grand Island, NY, USA) at 37℃ in an atmosphere of 5% CO2. Cells were grown until 90% confluence on a 6-well tissue culture plate and then starved for 18 hours. After that, the cells were treated with 400 µM cisplatin (Sigma, St. Louis, MO, USA) in phosphate buffered saline (control) for 8 hours. The cells were also treated with 1 µM PJ34 hydrochloride (a potent PARP1 inhibitor, R&D Systems, Minneapolis, MN, USA) in phosphate buffered saline (vehicle) at 2 hours before treatment with cisplatin.
SIRT3 knockout and wild-type cells
Cells were transfected with either SIRT3 Double Nickase Plasmid (catalog No. sc-425704-NIC) or Control Double Nickase Plasmid (catalog No. sc-437281) purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and then selected into SIRT3 knockout (KO) or wild-type (WT) using culture medium containing puromycin as modified from previous publication [26]. Briefly, cells were grown until 70% confluence on a 6-well tissue culture plate and then transfected with 3 g of plasmid DNA using 100 µl of UltraCruz Transfection Reagent (Santa Cruz Biotechnology). After 24 hours of incubation, cells were selected with culture medium containing 5 µg/ml puromycin antibiotic (Sigma), resulting the death of all non-transfected cells within 36 hours. Transfected cells were confirmed under fluorescent microscopy from 24 to 72 hours after puromycin selection. After starvation for 18 hours, cells underwent treatment with 1 µM PJ34 hydrochloride or vehicle for 2 hours followed by treatment with 400 µM cisplatin for 8 hours.
Western blot and immunoprecipitation
For western blotting, electrophoresis of protein extracts obtained from whole cell lysates using tris-glycine buffer systems and subsequent blotting was performed as previously described [10]. Membranes were incubated with antibodies against MnSOD (catalog No. 24127-1-AP, Proteintech, Chicago, IL, USA), copper/zinc superoxide dismutase (CuZnSOD; catalog No. 10269-1-AP, Proteintech), catalase (catalog No. 21260-1-AP, Proteintech), GPX (catalog No. SAB2700534, Sigma), β-actin (catalog No. A2228, Sigma), acetyl lysine (catalog No. MA1-2021, Thermo Fisher Scientific, Rockford, IL, USA), SIRT3 (catalog No. 10099-1-AP, Proteintech), and green fluorescent protein (catalog No. 66002-1-Ig, Proteintech). Peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) were applied, and a chemiluminescence reagent (PerkinElmer, Boston, MA, USA) was used to detect protein. The antiβ-actin antibody was used as a loading control on stripped membranes. Bands were quantified using Lab Works analysis software (Ultra-Violet Products, Cambridge, UK). For immunoprecipitation, cell lysates were mixed with the indicated antibody at 4℃ overnight. After this, protein A/G plus agarose (Santa Cruz Biotechnology) were added and incubated for 3 hours. After immune complexes were washed with lysis buffer, samples were boiled with sodium dodecyl sulfate reducing sample buffer and subjected to western blotting.
Enzyme activity
Mitochondria and cytosol were separated by differential centrifugation [27]. Superoxide dismutase activity was then measured from the mitochondrial and cytosol fractions respectively using a superoxide dismutase assay kit according to the manufacturer's instructions (catalog No. 706002, Cayman, Ann Arbor, MI, USA). Catalase activity in whole cell lysates was measured using a decomposition of hydrogen peroxide that was represented by a decrease in absorbance at 240 nm as previously described [28]. GPX and SIRT3 activities were measured in whole cell lysates using kits (catalog No. 703102 and 10011566, Cayman).
Forkhead box O3a transcriptional activity
For the analysis of the forkhead box O3a (FOXO3a) transcriptional activity, the SIRT3 KO and WT cells were infected with 1×106 plaque-forming units of adenovirus expressing wild-type FOXO3a (catalog No. 1576, Vector Biolabs, Eagleville, PA, USA) and then transfected with 10 µg FHRE-Luc (catalog No. 1789, Addgene, Cambridge, MA, USA), a luciferase construct generated by cloning three copies of forkhead response elements into pGL3-basic, using FuGENE 6 (Promega, Madison, WI, USA). After that, cells underwent 2 hours of treatment with 1 µM PJ34 or vehicle followed by 8 hours of treatment with 400 µM cisplatin. Luciferase activity was assayed using Bright-Glo Luciferase Assay System (Promega) according to the manufacturer's protocol.
LPO, 8-hydroxy-2'deoxyguanosine, and NAD+
LPO and 8-hydroxy-2'deoxyguanosine (8-OHdG) assays were performed in whole cell extracts using a LPO assay kit and a DNA/RNA oxidative damage ELISA kit (Cayman) according to the manufacturer's instructions, respectively. Total level of NAD+ was measured using a NAD+/NADH quantification colorimetric kit (BioVision, Mountain View, CA, USA) according to the manufacturer's instruction.
Mitochondrial membrane potential
The mitochondrial membrane potential was measured in HK2 cells seeded at a density of 105 cells per well on a 24-well plate, as previously described [29]. Briefly, tetramethylrhodamine, ethyl ester (TMRE; Abcam, Cambridge, MA, USA) (20 nM) was added to the cells and incubated for 30 minutes. After washing three times with 500 ml of phsphate buffered saline/0.2% fetal bovine serum three-times, the cells were read using a FilterMax F3 multimode microplate reader (Molecular Devices, Sunnyvale, CA, USA) at excitation and emission wavelengths of 549 nm and 575 nm, respectively.
Propidium iodide stain
The cells were stained with 1 µg/ml propidium iodide (PI; Sigma) for 10 minutes. After that, the percentage of PI-positive cells was assessed by fluorescence microscopy as previously described [10].
Results
PARP1 inhibition restores antioxidant defense system in cisplatin-induced injury to kidney proximal tubular cells
As recently demonstrated in a study by our group [15], cisplatin increases the concentrations of lipid hydroperoxide and 8-OHdG in an in vitro model of cisplatin nephrotoxicity, but PARP1 inhibition markedly attenuates the increase in oxidative stress. Subsequently, here we assessed whether the expression and activity of antioxidant enzymes could be diminished by PARP1 activation in the same model. In human kidney proximal tubule epithelial cells, cisplatin exposure for 8 hours decreased the expression of antioxidant enzymes including MnSOD, CuZnSOD, catalase, and GPX (Fig. 1A, B). However, the downregulation of MnSOD and catalase in those antioxidant enzymes was significantly attenuated following PARP1 inhibition (Fig. 1A, B). In addition to the decrease in expression, the activity of the antioxidant enzymes was decreased by exposure to cisplatin (Fig. 1C). In contrast, PARP1 inhibition significantly restored the activity of MnSOD, catalase, and GPX (Fig. 1C). This data indicates that PARP1 inhibition restores expression levels of MnSOD and catalase, and activity levels of GPX, MnSOD, and catalase in cisplatin-induced injury to human kidney proximal tubule epithelial cells.
PARP1 inhibition reduces acetylation of antioxidant enzymes in cisplatin-induced injury to kidney proximal tubular cells
Intriguingly, PARP1 inhibition reduced the decrease in GPX activity but not its expression. Furthermore, the activities of MnSOD, catalase, and GPX showed more dramatic decreases (<8% vs. vehicle+control) in cisplatin-injured cells, compared to the decreases in the expressions of those enzymes (<37% vs. vehicle+control). This severe alteration in enzyme activity can be implicated in the conformational change induced by the acetylation of lysine residues near its active sites [30]. To test whether PARP1 activation induces the acetylation of antioxidant enzymes during cisplatin nephrotoxicity, we performed immunoprecipitation using antibodies against MnSOD, CuZnSOD, catalase, and GPX enzymes in human kidney proximal tubule epithelial cells, and western blot analysis using an anti-acetyl lysine antibody. The acetylation level was measured using a ratio of the quantity of acetyl lysine to enzyme expression. This experiment showed that cisplatin exposure markedly increased acetylation of MnSOD, catalase, and GPX, while PARP1 inhibition significantly reduced such acetylation (Fig. 2A, B). Acetylation of CuZnSOD was not detected in either group (Fig. 2A). This data indicates that PARP1 activation triggers the acetylation of MnSOD, catalase and GPX in cisplatin-induced injury to human kidney proximal tubule epithelial cells.
PARP1 inhibition prevents SIRT3 downregulation induced by cisplatin in kidney proximal tubular cells
The SIRT3 enzyme is a stress-responsive deacetylase that protects against oxidative stress through the activation of MnSOD, catalase, and FOXO3-dependent antioxidant enzymes in various disease models [222331]. It has been reported that excessive PARP1 activation downregulates SIRT3 in endothelial cells under hyperglycemia [25]. Here, we assayed whether cisplatin-induced PARP1 overactivation decreases SIRT3 expression in human kidney proximal tubule epithelial cells using western blot analysis. As shown in Fig. 3A, SIRT3 expression was markedly decreased by cisplatin exposure, but its downregulation was completely prevented after PARP1 inhibition. Consistent with the observed expression pattern, cisplatin exposure significantly decreased SIRT3 activity in cells, but its activity was completely restored by PARP1 inhibition (Fig. 3B). Since both PARP1 and SIRT3 use the same cofactor, NAD+ [632], and further, PAPR1 activation influences SIRT3 activity through the modulation of NAD+ [33]; we additionally evaluated total level of NAD+ in all of groups. Cisplatin exposure dramatically decreased the total level, but PARP1 inhibition prevented this decrease (Fig. 3C). These experiments show that SIRT3 expression is regulated by PARP1 during cisplatin nephrotoxicity.
PARP1 inhibition improves activity of antioxidant enzymes through SIRT3-dependent deacetylation in cisplatin-induced injury to kidney proximal tubular cells
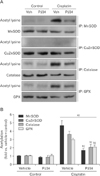
The improvement of antioxidant enzyme activity appears to be involved in SIRT3 upregulation through PARP1 inhibition during cisplatin nephrotoxicity. In order to confirm this, we made SIRT3-deficient kidney proximal tubule epithelial cells using transfection with double nickase plasmid encoding SIRT3 and treatment with a puromycin antibiotic (Fig. 4A), and evaluated the acetylation of antioxidant enzymes in cisplatin injury to SIRT3-deficient cells after PARP1 inhibition. In WT cells, PARP1 inhibition significantly reduced cisplatin-induced acetylation of MnSOD, catalase, and GPX (Fig. 4B, C). However, SIRT3 deficiency increased the acetylation of those enzymes after PARP1 inhibition, similar to those treated with vehicle (Fig. 4B, C), indicating that SIRT3 deficiency removes the deacetylative effect of PARP1 inhibition on MnSOD, catalase and GPX enzymes during cisplatin nephrotoxicity. Because of the direct effect of lysine acetylation on enzyme activity [30], we next assayed the activity of MnSOD, catalase, and GPX enzymes in cisplatin injury to SIRT3-KO and -WT cells. In cisplatin-injured WT cells, PARP1 inhibition increased the activity of MnSOD, catalase and GPX, while SIRT3-KO cells showed significant decreases in those enzyme activities after treatment with either PJ34 or vehicle (Fig. 4D). These results suggest that PARP1 inhibition prevents cisplatin-induced decreases in the activity of MnSOD, catalase, and GPX through SIRT3-dependent deacetylation.
PARP1 inhibition improves expression of antioxidant enzymes through SIRT3/FOXO3a axis in cisplatin-induced injury to kidney proximal tubular cells
We investigated the expression of those enzymes in cisplatin injury to SIRT3-KO and -WT cells after treatment with either PJ34 or vehicle. WT cells showed that PARP1 inhibition markedly increased MnSOD and catalase expression during cisplatin injury (Fig. 5A, B), but SIRT3 deficiency abolished the upregulation of both enzymes induced by PARP1 inhibition in cisplatin-injured cells (Fig. 5A, B). This data suggests that PARP1 inhibition restores MnSOD and catalase expressions through SIRT3 during cisplatin nephrotoxicity. The expression pattern of GPX in cisplatin-injured cells was significantly altered by PARP1 inhibition and/or SIRT3 deficiency (Fig. 5A, B), indicating that GPX gene expression is not involved in the protective effect of PARP1 inhibition against oxidative stress induced by cisplatin nephrotoxicity.
Subtypes of a FOXO transcriptional factor family have been shown to inhibit oxidative stress by enhancing the transactivation of antioxidant enzymes [2334]. Especially since FOXO3a has been found to upregulate MnSOD and catalase during stress [2235], we postulated that SIRT3 might have the ability to upregulate them by enhancing FOXO3a activity in cisplatin-injured cells. In order to test this hypothesis, we examined the transcriptional activity of FOXO3a in SIRT3-KO and -WT kidney proximal tubule epithelial cells during cisplatin injury after treatment with either PJ34 or vehicle. PARP1 inhibition markedly increased FOXO3a transcriptional activity in cisplatin-injured WT cells, but SIRT3 deficiency significantly prevented this increase (Fig. 6). After treatment with vehicle, no significant difference in its activity was observed between SIRT3-KO and -WT groups during cisplatin injury (Fig. 6). These results support the hypothesis that PARP1 inhibition enhances FOXO3a transcriptional activity through SIRT3, which may in turn transactivate MnSOD and catalase.
PARP1 inhibition is protective against oxidative stress and necrosis through SIRT3 in cisplatin-induced injury to kidney proximal tubular cells
Further to the results above, we postulated that the protective effect of PARP1 inhibition against oxidative stress was mediated by SIRT3 in cisplatin-injured kidney proximal tubular cells. In order to test this hypothesis, we assessed cisplatin-induced lipid hydroperoxide and 8-OHdG in SIRT3-KO and -WT cells after treatment with either PJ34 or vehicle. Consistent with our previous report [13], WT cells showed the decrease in lipid hydroperoxide and 8-OHdG concentration following PARP1 inhibition (Fig. 7A, B). However, SIRT3 deficiency significantly increased their concentrations following PARP1 inhibition as with the vehicle treatment (Fig. 7A, B), suggesting that SIRT3 deficiency completely removes the antioxidant effect of PARP1 inhibition in cisplatin-injured cells. Since the opening of mitochondrial permeability transition pore can be caused by oxidative stress in mitochondria, we additionally assessed the mitochondrial membrane potential using TMRE assay. In cisplatin-injured WT cells, PARP1 inhibition increased the mitochondrial membrane potential (Fig. 7C). However, SIRT3-KO cells revealed the significant decrease in the mitochondrial membrane potential regardless of treatment with either vehicle or PJ34 (Fig. 7C), suggesting that the antioxidant effect of PARP1 inhibition in mitochondria is implicated in SIRT3. Finally, to test whether PARP1 inhibition attenuates the necrotic cell death induced by cisplatin through SIRT3, we measured the percentage of necrosis in cisplatin-injured SIRT3-KO or -WT cells after treatment with either PJ34 or vehicle using a PI stain. As expected, PARP1 inhibition suppressed cisplatin-induced necrosis in WT cells (Fig. 7D); however, SIRT3 deficiency completely removed the suppressive effect of PARP1 inhibition in cisplatin-injured cells (Fig. 7D). Furthermore, cisplatin-induced necrosis in SIRT3-KO cells treated with vehicle was more severe, compared with that observed in WT cells treated with vehicle (Fig. 7D). These results support the hypothesis that PARP1 inhibition is protective against oxidative stress and necrosis during nephrotoxicity, acting through SIRT3.
Discussion
PARP1 activation is triggered by oxidative and nitrative stress-induced DNA damage, which is involved in the transactivation of various proinflammatory gene, and thus in the consequent inflammatory response [36]. As a result, recent studies have emphasized that PARP1 inhibition attenuates adhesion molecule expression, inflammatory cell infiltration, and the consequent secondary oxidative injury in acute kidney injury models [363738]. Here, we have identified that in cisplatin-induced nephrotoxicity in vitro using human kidney proximal tubular cells, (1) PARP1 inhibition restores the expression and activity of SIRT3; (2) PARP1 inhibition restores the activity of MnSOD, catalase, and GPX antioxidant enzymes through SIRT3-dependent deacetylation; (3) PARP1 inhibition attenuated the expression of MnSOD and catalase through SIRT3/FOXO3a axis-dependent transactivation; and (4) PARP1 inhibition blocked oxidative stress through SIRT3, thereby limiting necrotic cell death. Our data suggests that PARP1 inhibition plays a primary antioxidant role by activating SIRT3 during cisplatin nephrotoxicity, supported by our previous finding that PARP1 inhibition suppresses lipid peroxidation and oxidative DNA damage in cisplatin-injured human kidney proximal tubular cells [15].
SIRT3, the major mitochondrial deacetylase, is highly expressed in metabolically active tissues, such as kidney, liver, and heart [32]. Consistent with our results, its expression level has been shown to be decreased during cisplatin nephrotoxicity [24]. Intriguingly, we have first reported that PARP1 inhibition prevented the decrease in SIRT3 expression and activity under stress. As both PARP1 and SIRT3 use the same cofactor, NAD+ [632], PAPR1 activation influences SIRT3 activity through the modulation of NAD+ [33]. In contrast, the mechanism by which SIRT3 expression is upregulated by PARP1 inhibition has not been explored; however, based on a previous study of SIRT3, it can be presumed that improved energy deprivation by PARP1 inhibition under stress may contribute to the upregulated level of SIRT3 expression. It has been reported that, when AMP-activated protein kinase, which acts as a sensor of energy status is activated by treatment with its agonist, reduced SIRT3 expression in injured proximal tubular cells is significantly recovered [24]. There is an alternative mechanism by which PARP1 can also regulate SIRT3 expression at the level of gene transcription and posttranscription [2432]. PARP1 interacts with a number of transcription factors that regulate mitochondrial genes, and the activation of PARP1 represses their transcriptional activity through the poly ADP-ribosylation of the factors and/or their cofactors [639]. The further elucidation of the detailed mechanism will need additional investigation under our experimental conditions.
Whatever the mechanism of PARP1-regulated SIRT3 expression may be, our data herein has demonstrated that SIRT3 deficiency removes the antioxidative effect of PARP1 inhibition through the repression of the antioxidant defense system. MnSOD, which catalyzes the conversion of superoxide to hydrogen peroxide and oxygen, and catalase, which catalyzes the decomposition of hydrogen peroxide to water and oxygen, are critical antioxidant enzymes in the defense against oxidative stress. The expression levels of both MnSOD and catalase have been shown to be controlled by the FOXO3a transcription factor [40]; and its deacetylation mediated by SIRT3 upregulates those antioxidant enzymes and reduces oxidative stress, which further ameliorates acute kidney injury in mice [41]. In this study, we have demonstrated that PARP1 inhibition increases FOXO3a transcriptional activity and the expression level of both MnSOD and catalase in cisplatin-injured cells. Furthermore, SIRT3 deficiency eliminated those increases by PARP1 inhibition, suggesting that PARP1 inhibition upregulates both MnSOD and catalase through the SIRT3/FOXO3a axis during cisplatin nephrotoxicity. Other recent studies have reported that SIRT3 directly deacetylates and activates various antioxidant enzymes including MnSOD [31], catalase [18], and GPX [19]. Our present data have shown that PARP1 inhibition reduces the acetylation levels of MnSOD, catalase, and GPX through SIRT3 in cisplatin-injured cells; and then increases these enzyme activities. Notably, the patterns of acetylation and activity of GPX were observed without the alteration of GPX expression; suggesting that PARP1 inhibition activates the GPX enzyme through the deacetylation of SIRT3 without the transactivating process. This finding may also cause conflict between GPX activity and expression, and the dramatic alteration in the antioxidant enzyme activities. As has been previously reported, cisplatin exposure decreases CuZnSOD activity and expression in kidney proximal tubular cells [4243]; and, consistent with this, our data show decreased CuZnSOD activity and expression in cisplatin-injured kidney proximal tubular cells. However, its activity and expression were not altered by PARP1 inhibition, indicating no relationship between CuZnSOD and PARP1, at least within kidney parenchymal cells.
Oxidative stress is a critical contributor to cisplatin nephrotoxicity [44]. Cisplatin can induce either mitochondrial dysfunction, depletion of antioxidants, or cytochrome P450 system; and subsequently increase various reactive oxygen species (ROS) [454647]. ROS have a wide range of targets and modify multiple intracellular molecules, such as proteins, lipids, and DNA, resulting in severe injury. As such, several antioxidant enzymes have been investigated as potential compounds able to protect against cisplatin nephrotoxicity. For example, MnSOD overexpression and treatment with a SOD mimetic molecule have been shown to attenuate cisplatin-induced kidney tubular injury through suppression of ROS generation [4849]. Roles of SIRT3 and FOXO3a in the regulation of cellular ROS levels have been previously documented [4050]. In retinal capillary endothelial cells, PARP1-regulated SIRT3 ameliorates hyperglycemia-induced oxidative injury through the deacetylation of MnSOD [25]. In mouse hearts, SIRT3 prevents cardiac hypertrophy through FOXO3a-dependent antioxidant defense mechanisms [22]. In our previous studies, pharmacological and genetic inhibition of PARP1 suppresses cisplatin-induced oxidative stress in cultured kidney proximal tubular cells and in vivo kidney tissues [1315]. Those results from other and our previous studies are consistent with the present finding that PARP1 inhibition restores the activity and/or expression of antioxidant enzymes, SIRT3 and FOXO3a during cisplatin nephrotoxicity; and subsequently prevents ROS generation.
Taken together, this study demonstrates that cisplatin-induced PARP1 overactivation contributes to oxidative stress in kidney proximal tubule epithelial cells, and further, the increased oxidative stress is caused by enzyme dysfunction in antioxidant defense systems through the downregulation of SIRT3 signaling. PARP inhibition has potential to be an effective strategy for improving the outcomes of oxidative stress.






 XML Download
XML Download