

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The glutamate is an important excitatory neurotransmitter and its concentration is strictly controlled in the brain [1]. But excessive release of glutamate into extracellular leads to over-activation of glutamate receptors and induces neuronal damage [23]. Over-activation of glutamate receptors known Biologyas excitotoxicity plays an important role in many brain disorders including ischemic stroke, epilepsy, Alzheimer's disease, and Parkinson's disease [456].
Excitotoxicity is caused by excessive calcium influx into cells via glutamate receptors such as N-methyl-D-aspartate receptor (NMDAR) and induces calcium-dependent neuronal death [67]. Excessive activation of NMDAR promotes activation of calcium-dependent proteases including calpain and caspases that act as a signaling of cell death [8]. The calpain, neuronal proteases, subsequently triggers its-mediated cleavage of striatal-enriched tyrosine phosphatase (STEP) [910].
STEP exists two major isoforms of membrane associated STEP61 and cytosolic STEP46 and STEP61 is proteolytically cleaved by activated calpain, leading to production of STEP33 [11]. STEP regulate important synaptic signaling proteins including ERK1/2, p38 mitogen-activated protein kinase (MAPK), Fyn, and the NMDAR GluN2B subunit [111213]. Sustained activation of NMDAR GluN2B subunit cause significant degradation of active STEP, thus STEP serves as a modulator of NMDAR-dependent neuronal injury [9].
The hooks and stems of dried Uncaria sinensis has been traditionally used as a pharmacological medicine for treatment to diverse neurological symptoms. U. sinesis contains various classes of bioactive compounds such as caffeic acid, epicathchin, catechin and rhynchophylline exerting strong neuroprotective effects [14151617]. Our previous studies demonstrated that a hexane extract from U. sinensis exerts antiapoptotic effects against glutamate-induced neurotoxicity [18] and protective effects against cerebral ischemic damage [19].
In the further screening for active constituents using a neuronal cells and ischemic animal model, we isolated two specific fraction 6 (JGH6-7) and 43 (JGH43IA) among 62 fractions of hexane extract that shows most prominent neuroprotective effects. We reported the structure and neuroprotective effects of fraction 6 as a novel neuroprotective compound, 1-methoxyoctadecan-1-ol [20], but JGH43IA have not been investigated with regard to the cell death and its related signaling pathway. In the present study, we investigated the molecular mechanism underlying neuroprotective effects of fraction, JGH43IA, against glutamate-induced toxicity focusing on NMDAR and endogenous protease calpain with related STEP in primary cultured rat cortical cells.
Materials and Methods
Preparation of JGH43IA
Dried hooks and stems of U. sinensis were purchased from Hwalim Natural Drug (Busan, Korea) in September 2010. A voucher specimen (accession number PDRLCW-1) has been deposited in the Plant Drug Research Laboratory of Pusan National University (Miryang, Korea). The dried hooks and stems of U. sinensis (2.0 kg) were ground to a fine powder and then successively extracted at room temperature with n-hexane, ethyl acetate and methanol. Briefly, hexane extracts of U. sinensis were filtered and evaporated under reduced pressure at 45℃ and then lyophilized, which yielded a white powder of hexane extract (14.54 g). The hexane extract (11.31 g) was subjected to chromatography on a silica gel (40 µm; J.T. Baker, Phillipsburg, NJ, USA) column (70×8.0 cm) with a step gradient of 50% CHCl2 in hexane, 100% CHCl2, 5% and 20% acetone in CHCl2 and 5%, 25% and 50% MeOH in CHCl3 to obtain 62 fractions. Of these extracts, the solid form of faction 43 (23.1 mg, JGH43IA) was dissolved with dimethyl sulfoxide for further experiments.
Chemicals and antibodies
Cytosine-β-D-arabino furanoside, Hochest 33342, L-glutamate, 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), poly-L-lysine, and β-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). Neurobasal medium, B27, Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and other neuronal cell culture reagents were purchased from Gibco/Invitrogen (Carlsbad, CA, USA). For our investigations, the following antibodies were used: α-fodrin, calpain 1, calpain 2, CREB, phospho-CREB (pCREB, ser133), phospho-GluN2A (pGluN2A, Tyr1246) and phospho-GluN2B (pGluN2B, ser1303) were purchased from Cell Signaling Technology (Danvers, MA, USA). GluN2B and phospho-STEP (pSTEP, ser221/ser49) were purchased from Millipore (Billerica, MA, USA). GluN2A antibody was purchased from Upstate Biotechnology (Lake Placid, NY, USA). STEP antibody was purchased from Novus Biologicals (Littleton, CO, USA). Secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). FITC Annexin V apoptosis dectection kit was purchased from BD Bioscience (San Diego, CA, USA). A lactate dehydreogenase (LDH) cytotoxicity assay kit and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay kit were purchased from Promega (Madison, WI, USA).
Primary cortical neuronal culture
All procedures used in these studies followed the guidelines of protocols approved by the Pusan National University Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines. Time-pregnant Sprague-Dawley rats (E18-19) (Dooyeol Biotech, Seoul, Korea) were anesthetized and sacrificed by cervical dislocation. The cerebral cortex was carefully dissected out and transferred in ice cold stage; tissues were gently minced using a sterile razor blade and digested in 0.25% trypsin for 15 minutes in Ca2+ and Mg+-free Hanks' balanced salt solution, followed by mechanical dissociation using a pasteur pipette. After centrifugation, cells were re-suspended in DMEM supplemented with 10% FBS, 1 mM pyruvate, 4.2 mM sodium biscarbonate, 20 mM HEPES, 0.3 g/l bovine serum albumin, and 1% penicillin/streptomycin at a density of 1×10 cells/ml and grown on poly-L-Lysine-coated plates in a 5% CO2 humidified incubator at 37℃. After 48 hours, β-D-arabinofuranoside (5 µM) was added in order to prevent proliferation of non-neuronal cells. After 3 days in culture, the medium was exchanged to neurobasal medium containing 2% B27 supplement, 0.5 mM glutamine, and 1% penicillin/streptomycin. Cortical neurons were maintained in primary culture for 9-10 days for use in experiments.
MTT assay
The MTT assay was used for determination of cell viability. Cortical neurons were cultured at a density of 5×104/well in 96 well plates and treated with JGH43IA at different concentration for 24 hours, after treatment with 200 µM of glutamate for 6 hours, the medium was replaced with 0.5 mg/ml MTT solution for 4 hours at 37℃. After incubation, formazan crystals produced in cells were solubilized in dimethyl sulfoxide. Absorbance was measured at 570 nm using a Spectra MAX 190 spectrometer (Molecular Devices, Sunnyvale, CA, USA). Results were expressed as a percentage of control.
LDH assay
Measurement of LDH released from damaged neurons was performed for assessment of cytotoxicity. After treatment, cortical neurons were lysed by addition of lysis solution, followed by incubation at 37℃ for 45-60 minutes. Supernatant samples were transferred to a 96 well enzymatic assay plate, followed by addition of substrate to each sample. The enzymatic reaction was allowed to proceed for 30 minutes at room temperature, protected from light. After the enzymatic reaction was stopped, the plate was read at 490 nm using a Spectra MAX 190 spectrometer (Molecular Devices). Data represent the percentage of LDH released relative to controls.
Hoechst staining
Cortical neurons were grown on Lab-Tek chamber slides (Thermo Fisher Scientific Inc., Rochester, NY, USA) at a density of 1×105 cells/ml. After treatment, the cells were fixed in 4% paraformalehyde for 20 minutes at 4℃. Fixed cells were washed three times with phosphate buffered saline (PBS), followed by staining with 10 mg/ml Hoechst 33342 in PBS for 10 minutes at 37℃. The cells were washed three times with PBS and mounted using the medium for fluorescence (Vector Laboratories Inc., Burlingame, CA, USA), followed by observation under a laser scanning confocal microscope (LSM 510, Carl Zeiss, Oberkohen, Germany). Data are presented as the ratio of chromosomal condensation and morphological change as a percentage of total cells.
TUNEL assay
Cortical neurons were grown on Lab-Tek chamber slides (Thermo Fisher Scientific Inc.) at a density of 1×105 cells/ml. After treatment, the cells were fixed in 4% methanol-free formaldehyde solution in PBS (pH 7.4) for 25 minutes at 4℃. Fixed cells were then washed with PBS, followed by incubation with DNA-labeling solution at 37℃ for 60 minutes inside a humidified chamber to allow the tailing reaction to occur. The DNA-labeling reaction was terminated by addition of 2× SSC, followed by washing for removal of unincorporated fluorescein-12-dUTP. The cells were then counterstained with 1 mg/ml propidium iodide (PI) solution in PBS for 15 minutes at room temperature in the dark. Apoptotic neurons were determined by localization of green fluorescent cells (fluorescein-12-dUTP) on a red background using a fluorescence microscope (Carl Zeiss). Data are presented as apoptotic cells as a percentage of total cells.
Flow cytometry analysis
After treatment, cortical neurons were harvested and cells were washed twice with cold PBS, followed by re-suspension in binding buffer at a concentration of 1×105 cells/ml; 100 ml of the solution were transferred to a flow cytometric tube, followed by double staining with Annexin V-FITC and PI in the dark at room temperature for 15 minutes. Subsequently, 400 ml of binding buffer was added and analysis of samples was performed using a flow cytometer (FACS CantoTM II, Becton Dickinson, San Jose, CA, USA).
Western blot
After treatment, cortical neurons were washed in cold PBS buffer, followed by homogenization in lysis buffer (200 mM Tris [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1 mM NaF, 1% NP40, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3Vo4, and protease inhibitor cocktail). Equal amounts of proteins were then separated by 10%-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis; the resolved proteins were then transferred to a nitrocellulose membrane (Whatman, Dassel, Germany). The membrane was blocked with 5% skim milk, followed by exposure to the appropriate antibodies. Using an enhanced chemiluminescence system (Pierce Biotech, Rockford, IL, USA), all bands were visualized using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Results of Western blot assay reported here are representative of at least three experiments.
Data analysis
All data were expressed as mean±SEM and the SigmaStat statistical program version 11.2 (Systat Software, San Jose, CA, USA) was used for data analysis. The paired student's t test was used for analysis of data. A value of P<0.05 was considered to indicate a statistically significant result.
Results
Treatment with JGH43IA reduces glutamate-induced neuronal toxicity
We investigated the protective effects of JGH43IA from U. sinensis against glutamate-induced toxicity using MTT and LDH assay. Treatment with glutamate alone led to reduce in cell viability to approximately 68% of the control group, but pretreatment with JGH43IA exhibited in a significant gradual decrease of glutamate-induced toxicity in a dose-dependent manner (Fig. 1A). These data were further verified by the LDH assay. The level of LDH release increased to approximately 58% after exposure to glutamate. However, pretreatment with JGH43IA showed a significant decrease of this release in a dose-dependent manner (Fig. 1B). These results suggest that JGH43IA pretreatment has a significant protective effect against glutamate-induced toxicity in primary cortical neurons.
Treatment with JGH43IA inhibits glutamate-induced apoptotic neuronal death
We performed Hoechst 33342 staining and TUNEL assay in order to characterize the types of neuronal death by JGH43IA treatment. Results of Hoechst staining indicated that treatment with glutamate alone exhibited high levels of condensed chromatin and apoptotic bodies. But pretreatment with JGH43IA showed a significant decrease of these apoptotic features at both concentrations of 0.01 and 0.1 µg/ml (Fig. 2). DNA fragmentation was also evaluated by TUNEL assay as another method for detection of apoptosis. TUNEL-positive cells increased to 29.6% in total cells after exposure to glutamate alone. However, pretreatment with JGH43IA resulted in significantly decreased of TUNEL-positive cells to nearly 19.7% and 9.4% at a concentration of 0.01 and 0.1 µg/ml, respectively (Fig. 3).
We further confirmed these results by use of flow cytometry using Annexin V FITC/PI staining. The percentage of apoptotic cells reached up to 21.4% in glutamate alone-treated neurons, whereas, the increase in necrotic cells was only 4.8%. But, treatment with JGH43IA showed a markedly decrease in the number of glutamate-induced cells death to 10.8% and 5.7% at a concentration of 0.01 and 0.1 µg/ml, respectively (Fig. 4). These results suggest that cortical neurons are to mainly undergo apoptotic neuronal death after exposure to glutamate and pretreatment with JGH43IA exerts a neuroprotective effect by abrogating apoptotic cell death.
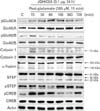
Treatment with JGH43IA down-regulates activation of NMDAR GluN2B subunit and calpain-mediated STEP cleavage
The NMDAR and calpain-mediated STEP play a dominant role in mediating the toxic signal under glutamate exposure in our previous study [20]. Thus we attempted to determine whether JGH43IA could inhibit both activation of NMDAR and calpain and subsequent STEP cleavage caused by glutamate. Our previous study shows that NMDAR GluN2A and GluN2B subunit shows a rapid increase in expression of phosphorylation at early time points with a rapid increase in the active form of calpain 1 (~75 kDa), subsequently resulting in a marked increase in a breakdown product of fodrin (~145/150 kDa), indicative of cleavage by activated calpain. The STEP, substrate for Ca2+-dependent calpain, is also rapidly cleaved to a smaller isoform, STEP33 following exposure to glutamate alone. Pretreatment with JGH43IA showed a marked selective decrease in glutamate-stimulated phosphorylation of the NMDAR GluN2B subunit. Pretreatment with JGH43IA reduced calpain1 activity and also reduced the accumulation of fodrin breakdown product caused by glutamate with significant blockage of STEP33 production. Especially, pretreatement of JGH43IA resulted in reduced ~145 kDa fodrin production of 30.2% compared with glutamate-treated cells at 360 minutes after glutamate exposure. And lower subsequent STEP33 production, 28.4% compared with glutamate-treated cells, also observed at early phases after glutamate exposure in JGH43IA-treated cells [20]. Pretreatment with JGH43IA increased in the levels of phosphorylated CREB compared to glutamate alone treated cells (Fig. 5). These results suggest that inhibition of NMDAR GluN2B subunit activation and subsequent calpain-mediated STEP cleavage are involved in the neuroprotective mechanisms of JGH43IA.
Discussion
Several alkaloids and phenolic compounds from hooks and stems of U. sinensis have been reported to exert neuroprotection and provide relief from various nervous related symptoms [142122]. Especially, the main alkaloid constituent, rhynchophylline, shows strong antihypertensive and neuroprotective activities [22]. We isolated specific fraction 43 (JGH43IA) from hexane extract of U. sinensis that shows prominent effects in cell viability and neurological scores in neuronal cells and focal cerebral ischemia model to screen active constituents, respectively.
Thus we investigated on the neuroprotective effect of JGH43IA that may have potential therapeutic value in treatment of brain disorder. Results of this study showed protective effects of JGH43IA against glutamate-induced toxicity via reduction of apoptotic cell death in primary cortical neuronal cells. Pretreatment with JGH43IA resulted in attenuated activation of NMDAR GluN2B and active form of calpain 1 leading to subsequent cleavage of STEP. Together, these results strongly suggest that JGH43IA can act as a neuroprotection capable of down-regulating NMDAR GluN2B subunit and calpain linked to STEP cleavage against glutamate-induced neuronal toxicity.
Excitotoxicity by excess glutamate stimulation contributes to an imbalance in survival versus death signaling pathways in various regions of brain that caused neuronal dysfunction and degeneration [2324]. However, glutamate-induced neuronal death is depending on severity of stimulation and exposure time to determine death types, necrosis and apoptosis, or both, mixed type [2526]. First, we demonstrated that pretreatment with JGH43IA effectively protects impaired cortical neurons with glutamate stimulation and results mainly in protection against neuronal apoptosis, because cortical neurons undergo extensive apoptotic-like deaths under our experimental conditions.
In sequence, we investigated activation of NMDAR subunit and related calapin-mediated STEP cleavage to clarify underling molecular mechanisms of apoptosis. NMDARs leads to difference result of both neuroprotective and neurotoxic functions according to their located sites. Stimulation of synaptic and extrasynaptic NMDAR promotes the neuroprotection and cell death, respectively [27]. GluN2B-NMDAR is located primarily at the extrasynaptic site, this subunit is generally thought to be involved in triggering intracellular cascades that lead to neuronal apoptosis following an excitotoxic insult [928]. Furthermore, phosphorylation of the NMDAR GluN2B subunit at the 1303 site leads to ischemic neuronal death [29]. We simultaneously performed western assay of JGH43IA in glutamate-treated primary neuronal cells at same conditions with previous study to determine basic molecular mechanism of neuroprotection [20]. Our present western blot results showed that pretreatment with JGH43IA reduced phosphorylation of GluN2B-NMDAR at the same site suggesting GluN2B subunit could play a dominant role in arrest of neuronal death.
Excessive calcium influx via activation of NMDAR contributes to calcium-dependent neuronal death, triggering intracellular cascades [711]. The calcium-dependent protease calpains also require an increase in cytosolic concentration of calcium via extracellular influx in glutamate-treated cells [3031]. Extrasynaptic stimulation of NMDAR promotes selective calpain-mediated proteolysis of STEP61, producing the truncated cleavage product STEP33, and these cleavage activates p38 MAPK leading to excitotoxic neuronal injury [91132]. Therefore, STEP61 is a valid target for development of neuroprotective therapy, because of regulating important synaptic signaling proteins [911].
When we treated glutamate alone in primary cortical neuronal cells, our previous results are consistent with general glutamate-induced toxicity [2033]. The active form of calpain 1 (~75 kDa) was clearly observed following exposure to glutamate and activation of this protease was confirmed by α-fordrin cleavage (~150 kDa). Up-regulated activation of calpain by glutamate alone treatment also led to subsequent decrease of active form of STEP [20]. But pretreatment with JGH43IA showed an attenuated activation of calpain 1 with decreased α-fodrin fragment. Subsequently, JGH43IA pretreatment recovered level of STEP61 with reduced production of STEP33. Overstimulation NMDAR with glutamate exert their neurotoxic effects in part by overactivation of calpains, but number of contradictory studies such as neuroprotection or neurotoxicity have been performed to understand its roles in neurodegenerative diseases [34]. We found that inhibition of calpain I-dependent STEP cleavage was involved in JGH43IA-mediated neuroprotection in the present study.
Thus our results suggest that calpain 1 may be activated by significant increases in cytosolic Ca2+ via GluN2B-NMDAR and that JGH43IA can selectively attenuate these activations involving in triggering intracellular cascades for phosphorylation. Inhibition of calpain-mediated STEP cleavage by JGH43IA may inhibit chronic activation of ERK and p38 leads to activation of apoptotic pathways [35]. Activation of CREB leads to expression of many genes associated with cell survival by transcriptional activation, leading to expression of many genes associated with cell survival in neurodegenerative disorders [363738]. Activation of CREB was significantly increased by pretreatment with JGH43IA in the present study. Thus, neuroprotective effects of JGH43IA may be linked to activation of CREB leading to prevention of neuronal death.
Despite neuroprotective activities of JGH43IA, our study did not investigate its main functional components. For identification of therapeutic agent for brain disorder, further studies of functional components against glutamate-induced excitotoxicity will be needed. Consequently, JGH43IA exert neuroprotective effects against glutamate-induced apoptosis in primary cultured neurons by down-regulation of GluN2B-NMDAR and calpain activation with subsequent reduction in STEP cleavage. This single fraction from Uncaria sinensis may be a highly effective therapeutic agent in treatment of neurodegenerative diseases associated with excitotoxic insults.




 XML Download
XML Download