

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Angiogenesis is a process whereby new capillary blood vessels are produced from preexisting vessels. This process is controlled by interplay between growth factors, which either stimulate or inhibit blood vessel growth. Furthermore, some molecules, such as, vascular endothelial growth factor, basic fibroblast growth factor, and thymidine phosphorylase, have been reported to have angiogenic activities. Angiogenesis is crucial for normal processes, such as, fetal development, tissue repair, and the female reproductive cycle, and for abnormal processes, such as, tumor growth, arthritis, stroke, ulcer development, and diabetic retinopathy [1].
In view of the widespread use of caffeine in food and beverages, and in over-the-counter medications, such as, cold and flu remedies, any effect on angiogenesis could be of substantial importance [2, 3]. Furthermore, the caffeine molecule is structurally similar to adenosine, and it has been well established that caffeine acts as an adenosine receptor antagonist. Adenosine receptors belong to the G-protein-coupled 7 transmembrane super family of cell surface receptors, which are composed of A1, A2A, A2B, and A3 subtypes [4-8]. Adenosine, a potent endogenous physiological mediator, regulates many physiological processes, and it has been reported that adenosine and adenosine agonists up-regulate the expressions of angiogenic factors, down-regulate anti-angiogenic factor thrombospondin-1 (TSP-1) via adenosine receptors, and induce angiogenesis [9-15]. In addition to studies performed in vitro, numerous in vivo studies have shown that adenosine and adenosine agonists induce blood vessel growth in the corneas of amphibian, avian, and mammalian embryos and in skeletal muscle, the body, and chick chorioallantoic membrane (CAM) [15-24]. However, somewhat surprisingly, the effects of caffeine (a known adenosine receptor antagonist) on angiogenesis and on the expressions of anti-angiogenic factors have not been previously investigated.
TSP-1 is a naturally occurring inhibitor of angiogenesis that is synthesized and secreted by fibroblasts, endothelial cells, tumor cells, and others [25, 26]. TSP-1 has been shown to play a critical role in the inhibition of angiogenesis [27], and thereby, to inhibit tumor growth and experimental metastasis [25-31]. Furthermore, an inverse correlation has been reported between TSP-1 expression and microvessel density in vivo [32-34], and in another study, TSP-1 was found to mediate endothelial cell apoptosis [35, 36] and inhibit angiogenesis in association with the expressional down-regulation of Bcl-2 and the processing of caspase-3 into smaller proapoptotic forms [28].
In the present study, we examined the effects of caffeine on angiogenesis and on the expression of the anti-angiogenic factor TSP-1, and sought to identify the molecular mechanism responsible for its activities.
Materials and Methods
Drugs and reagents
The following drugs were used: caffeine, 5'-N-ethyl-carboxamino-adenosine (NECA), 2-p-(2-carboxyethyl) phenylethylamino-NECA (CGS21680), 4,6-diamino-2-phenylindole (DAPI), phenazine methosulfate (PMS), MTS (Sigma, ST. Louis, MO, USA); anti-actin (Delta Biolabs, Gilroy, CA, USA); anti-TSP-1 antibody (Lab Vision, Fremont, CA, USA); anti-caspase-3 antibody (Cell Signaling, Berverly, MA, USA); anti-Bcl-2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA); peroxidase labeled anti-mouse IgG (H+L) and peroxidase labeled anti-rabbit IgG (H+L) (Vector Laboratories Inc., Burlingame, CA, USA); YO-PRO-1 and Alexa fluor 594 labeled goat anti-rabbit IgG (H+L) (Molecular Probes, Eugene, OR, USA); Matrigel basement membrane matrix (BD Biosciences, Bilerica, MA, USA); protein extraction solution, western blot detection reagents (ELPIS, Daejeon, Korea); and fluorescent mounting medium Dako (Carpinteria, CA, USA). Protein assay kits were obtained from Bio-Rad (Hercules, CA, USA). All other chemicals were of reagent grade and were obtained commercially.
Chick CAM in vivo assay
The chick embryonic CAM assay was performed as previously described [37, 38]. Briefly, fertilized chicken eggs were incubated for 48 hours in a humid 38.5℃ atmosphere. After extracting 2-3 ml of albumin, a small window was opened in each egg in order to allow separation of CAM and shell during embryo development. Windows were temporarily sealed with adherent tape and eggs were incubated for a further 4 days. Drugs were prepared as previously described [39]. Methylcellulose solutions (2%) were mixed with equivalent volume of distilled water (without or with caffeine in a range of concentrations 250, 500, and 1,000 mM) in each group and placed 20 µl of each solution on the tips of 3.2-mm diameter Teflon rods and allowed to dry for 1 hour in laminar flow hood. Dried disks were removed from the Teflon rods and each disk was placed on each CAM. Windows were then sealed and eggs were incubated for additional 48 hours. Fat Emulsion (Intralipos, Fresenius Kabi, Warrington, UK) was then injected into chorionic cavity, and images of blood vessel images were captured under a stereomicroscope. Lengths of blood vessels in drug-treated regions were analyzed using the iMT Image Analysis program (iMTechnology, Bucheon, Korea).
Culture of human umbilical vein endothelial cells (HUVECs)
HUVECs were prepared as described previously [40, 41]. Briefly, HUVECs were harvested by collagenase treatment of umbilical cords obtained from the Department of Obstetrics at Chungbuk National University Hospital. The veins were cannulated, washed with Dulbecco's phosphate-buffered saline (PBS; GibcoBRL, Grand Island, NY, USA), filled with 0.2% collagenase (Sigma), and incubated for 15 minutes in a water bath at 37℃. The contents of each vein were collected and this was followed by an additional PBS wash to remove remaining cells. Cells were centrifuged at 1,100 rpm for 3 minutes and supernatant was discarded. The cell pellet was resuspended in M199 culture medium (GibcoBRL) containing 20% heat-inactivated fetal bovine serum (Biowhittaker, Walkersville, MD, USA) supplemented with 25 µg/ml endothelial cell growth factor (Biotechnology Inc., Stoughton, MA, USA), 2 mM L-glutamine (GibcoBRL), 100 µg/ml heparin (GibcoBRL), and 500 µg/ml antibiotic/antimycotic (GibcoBRL). Cells were cultured in HUVEC medium in a tissue culture flask coated with 1% gelatin (Sigma). Isolated HUVECs were characterized by their typical cobblestone morphology and stained for factor VIII-related antigen and CD31.
Proliferation assay
Endothelial cell proliferation was determined by using an MTS assay [42]. Cells were seeded onto gelatin-coated 96-well plates at a density of 5×103 cells/100 µl per well, cultured overnight, and treated with caffeine (0, 25, 50, 100, 250, or 500 µM) for 24, 48, or 72 hours. Cells were then rinsed with PBS, treated with phenol red-free M199 and MTS/PMS, and incubated for 4 hours. Absorbance was read at 492 nm using a microplate reader (Multiskan MS photometer type 352, Lab Systems, Helsinki, Finland).
Flow cytometry
The apoptosis induced by caffeine was investigated using the YO-PRO-1 Apoptosis Detection Kit (Molecular Probes). Briefly, at 24, 48, and 72 hours after adding caffeine (500 µM), cells were trypsinized, washed twice with ice-cold Dulbecco's PBS, and incubated for with YO-PRO-1 on ice 15 minutes. Cells were analyzed by flow cytometry using a Becton Dickenson FACScan flow cytometer and Cell Quest software ver. 1.2 (Becton Dickenson, San Jose, CA, USA) within 30 minutes. Non-treated cells were used as a positive control.
Western blot analysis
To determine protein contents, HUVECs were collected by scraping in 1× lysis buffer, and then sonicated three times for 5 seconds on ice. After centrifugation (14,000 rpm, 10 minutes, 4℃), supernatant was transferred to a new microcentrifuge tube. Protein concentration in supernatant was measured using a protein assay kit (Bio-Rad). Absorbance was measured using a spectrophotometer (UVIKON 860, Kontron Co., Zurich, Switzerland) at 595 nm.
TSP-1 expressions were determined by western blotting. Briefly, HUVECs were treated with caffeine, CGS-21680, or NECA for 12 hours, rinsed once with ice-cold PBS, scraped into 1× lysis buffer, and sonicated three times for 5 seconds on ice. After centrifugation (14,000 rpm, 10 minutes, 4℃), supernatant was transferred to a new microcentrifuge tube. Protein concentration in supernatant was measured using a protein assay kit (Bio-Rad). After inactivation at 95℃ for 5 minutes and cooling on ice for 5 minutes, samples were separated by 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were then blocked for 30 minutes at room temperature in blocking solution (5% non-fat dry milk in 1× Tris-buffered saline/Tween-20 [TBS-T]), incubated at 4℃ overnight on a rocker in blocking solution containing a 1:1,000 dilution of primary antibody for anti-angiogenic factor TSP-1, and then incubated with alkaline phosphatase-labeled anti-goat secondary antibody. Protein bands were visualized using an enhanced chemiluminescence (ECL) kit and quantified using Scion Image software.
Caspase-3 and Bcl-2 expression
HUVECs were treated with caffeine for 36 hours, and lysed in lysis buffer on ice. Cells were then scraped off, transferred to microcentrifuge tubes, and sonicated three times on ice. Soluble extracts of cells were prepared by centrifugation at 14,000 rpm for 15 minutes at 4℃. Protein concentrations were measured and then equal amounts of proteins (50 µg per sample) were mixed with 1× sample buffer. After inactivation at 95℃ for 5 minutes and cooling on ice for 5 minutes, samples were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes for 20 minutes at 50 V in transfer buffer (25 mM Tris base [pH 8.5], 0.2 M glycine, 20% methanol). Membranes were then blocked for 30 minutes at room temperature in blocking solution (5% non-fat dry milk in 1× TBS/T) on a rocker. For the western blotting of Bcl-2 and caspase-3, membranes were incubated with primary antibodies (anti-Bcl-2 and anti-caspase-3 antibody) diluted 1:1,000 in TBS/T containing 3% non-fat dried milk with agitation overnight at 4℃. After washing three times with TBS/T, membranes were incubated with alkaline phosphatase-labeled anti-goat secondary antibody. Protein bands were visualized using an ECL kit and quantified using Scion Image software.
Immunofluorescence staining
HUVECs were seeded on coverslips and allowed to adhere overnight, and then cells were treated with caffeine with or without NECA, CGS21680 for 12 hours. Adherent cells were washed with PBS three times and fixed for 10 minutes with 2% paraformaldehyde. After fixation, cells were washed twice with 0.1 M glycine-PBS for 10 minutes, blocked with 10% normal goat serum for 30 minutes at room temperature, incubated with polyclonal anti-TSP-1 antibody (NeoMarkers, Fremont, CA, USA) for 30 minutes at 37℃, washed three times, and labeled with 5 µg/ml Alexa Fluor 594-conjugated goat anti-rabbit IgG (Molecular Probes) for 30 minutes. After secondary antibody conjugation, cells were washed and counter stained with 2 µg/ml DAPI (Sigma) for 5 minutes at 37℃, washed three times, and mounted with fluorescent mounting medium (Dako). Stained cells were examined under a confocal laser scanning microscope (CLSM-2000, Leica, Wetzlar, Germany).
DAPI staining was performed as previously described. Briefly, cells were cultured on coverslips in 4-well dishes (Nalge Nunc International, Naperville, IL, USA), treated with caffeine (500 µM for 48 hours), collected, and fixed by incubation in cold methanol for 10 minutes. After washing in PBS, cells were incubated in 2 ng/ml DAPI solution for 30 minutes in the dark, and then observed under a fluorescence microscope (Zeiss, Oberkochen, Germany).
Statistical analysis
Results are shown as the means±SDs of at least three independent experiments performed in triplicate. The statistical significances of differences between means were determined using the Student's t-test for unpaired values in Sigmaplot 5.0. Statistical significance was accepted for P<0.05.
Results
Inhibitory effect of caffeine on angiogenesis
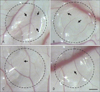
In vivo CAM assays were used to determine the effect of caffeine on angiogenesis. In the control group (treated with H2O), a normal branching pattern of blood vessels was present (Fig. 1A). However, the number of blood vessel branches was dose-dependently reduced by caffeine at 250, 500, or 1,000 µM for 48 hours (Fig. 1B-D). Compared the controls, relative lengths of branches were 84.7%, 66.6%, and 61.2% after caffeine treatment at 250, 500, and 1,000 µM, respectively (Fig. 2). These results indicate that caffeine dose-dependently inhibits the formation of new blood vessels in chick embryonic CAMs.
Effects of caffeine on the proliferation of endothelial cells
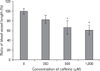
The effect of caffeine on HUVEC proliferation was investigated using an MTS assay. Proliferation was significantly and dose-dependently inhibited after treatment with caffeine for 72 hours. No significant differences versus the control group were observed at caffeine concentrations lower than 100 µM. However, at concentrations of 250 µM and 500 µM, caffeine inhibited HUVEC proliferation HUVECs to 83.4% and 39.5% of the control, respectively (Fig. 3). These findings indicated that caffeine dose-dependently inhibits HUVEC proliferation.
Effects of caffeine on HUVEC apoptosis
Flow cytometry was used to determine whether the inhibition of proliferation was due to apoptosis. The YO-PRO-1 kit was used to analyze percentages of apoptotic cells induced by caffeine. Yopro stains early apoptotic cells and propidium iodide stains late apoptotic cells. Caffeine (500 µM) induced apoptosis in a time-dependent manner. Percentages of total apoptotic cells were 19.2%, 32.4%, and 50.4% after treatment with 500 µM caffeine for 24, 48, and 72 hours, respectively. However, only 8.5% of cells in the control group were apoptotic (Fig. 4). These results show that at 500 µM caffeine inhibits HUVEC proliferation by inducing apoptosis in a time-dependent manner.
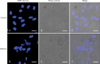
The apoptotic effect of caffeine on HUVECs was confirmed by examining morphological changes. Under the phase-contrast microscope, about 50% cells treated with caffeine at 500 µM for 72 hours were detached and exhibited cell rounding, cytoplasmic blebbing, and an irregular shape (data not shown). Furthermore, DAPI and differential interference contrast images obtained using a confocal laser scanning microscope showed that caffeine-treated HUVECs exhibited nuclear condensation and perinuclear apoptotic bodies (Fig. 5).
Effects of caffeine on the expression of TSP-1 in endothelial cells
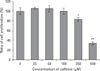
Because it has been previously reported that adenosine and adenosine receptor agonists decrease the expression of the anti-angiogenic protein TSP-1 [9-15], we examined the effect of caffeine on TSP-1 expression in HUVECs. Although TSP-1 expression was not affected by caffeine at 250 µM or less, it increased 1.5-fold after treatment with 500 µM caffeine for 12 hours (Fig. 6). In addition, the concentration of TSP-1 in culture medium increased dose-dependently after 24 hours of caffeine treatment. In addition, TSP-1 expression increased after only 1 hour of treatment with 500 µM of caffeine, and this increase was maintained until 24 hours (data not shown). These results indicate that caffeine increases the expression of TSP-1 in HUVECs in a dose-dependent manner.
Upregulation of TSP-1 via the inactivation of the adenosine A2B receptor
Based on a report that HUVECs express A2A and A2B adenosine receptors [9], we used the selective A2A agonist CGS2180 and the nonselective A2A/B agonist NECA to determine the subtypes of adenosine receptors involved in the up-regulation of TSP-1 in HUVECs. As compared with untreated controls, CGS21680 did not alter TSP-1 protein levels. However, NECA at 100 µM decreased TSP-1 protein levels to 78.2% of the control (Fig. 7). Immuno-fluorescence staining findings were in accord with Western blotting findings (Fig. 8). These results suggest that TSP-1 overexpression by caffeine is due to the inactivation of adenosine A2B receptors in HUVECs.
Regulation of caspase-3 and Bcl-2 expression by caffeine
To understand the mechanisms involved in caffeine-induced endothelial cell death, we examined the expressions of caspase-3 and Bcl-2. After adding 500 µM of caffeine to cell culture, over 50% of HUVECs underwent apoptosis after 72 hours (Figs. 3, 4). To determine whether caffeine induces apoptosis via the activation of caspase-3, we used a specific antibody for the pro-apoptotic form of caspase-3 was used. When HUVECs were treated with caffeine at 250 µM or 500 µM for 36 hours, caspase-3 expression increased to 191.3% and 305.8%, respectively (Fig. 9). In addition, treatment with 500 µM caffeine for 36 hours was found to halve the expression of anti-apoptotic Bcl-2 (Fig. 10). These results indicate that caffeine induces endothelial cell apoptosis by modulating the regulating the expressions of Bcl-2 and caspase-3.
Discussion
Previous studies have shown that caffeine inhibits carcinogenesis in animal models [43, 44]. However, although it was suggested that the mechanisms responsible for the anti-carcinogenic effects of caffeine are related to its inhibition of cancer cell proliferation [45-48] and to increasing cellular sensitivity to certain chemotherapeutic agents and penetrating radiation [43, 44, 49], it remains a possibility that the anti-carcinogenic effects of caffeine are due to its anti-angiogenic effects. The results of the CAM assay performed in the present study demonstrate for the first time that caffeine has an inhibitory effect on angiogenesis.
To explore the mechanisms responsible for this inhibition of angiogenesis by caffeine, we examined the effects of caffeine on endothelial cell proliferation, which are essential the first steps of angiogenesis. In the present study, we used HUVECs because they are human cells, and thus, the results obtained would be directly applicable to clinical research in man. Second, HUVECs are easily obtained from umbilical cords by primary culture. Third, because they are frequently used in vascular research, much basic data is available. The results of this study showed that caffeine inhibited HUVEC proliferation in vitro in a time- and dose-dependent manner via the induction of apoptosis (Figs. 3, 4). On the other hand, caffeine had no effect on HUVECs differentiation on Matrigel or migration in vitro (data not shown). These results indicate that inhibitory effect of caffeine on angiogenesis is related, at least in part, to its apoptotic effect [50, 51].
As compared to previous studies, the concentration of caffeine used in the study was low. However, despite the low concentrations used, the effect of caffeine was significant. Previous reports indicate that high concentrations of caffeine induce apoptosis in several cell lines, for example, at 10 mM in human neuroblastoma cells [52], 4 mM in human pancreatic adenocarcinoma cells [53], and at 5 mM in human A549 lung adenocarcinoma cells [54]. However, high concentrations of caffeine may not be relevant under physiological conditions because of its undesirable side effects. A recent study showed that a low concentration of caffeine (450 µM) induced the apoptosis of JB6 C141 cells, and in the present study, the maximum concentration used in the apoptosis investigation was 500 µM in cell culture. That is to say, caffeine at more less concentrations than other researches may induce endothelial cell apoptosis, and thus, inhibit angiogenesis.
It has been well established that angiogenesis is regulated by a balance between pro-angiogenic and anti-angiogenic factors, which are produced and secreted by various cell types. A recent study indicated that stimulation of adenosine receptor increases angiogenesis by down-regulating the production of TSP-1, an anti-angiogenic factor [15]. Thus, because caffeine is an adenosine receptor antagonist, we study investigated if it up-regulates TSP-1. Interestingly, we found that TSP-1 was expressed and higher levels and that its secretion to medium increased after treating HUVECs with caffeine (Fig. 6). These data suggest that the inhibitory effect of caffeine on angiogenesis is related to the up-regulation of TSP-1.
Angiogenic effects of adenosine receptors on endothelial cells remain debatable. Previous studies have indicated that adenosine A2B receptors enhance angiogenesis [9, 10, 55-57], whereas other studies using models of wound healing in A2A knockout mice showed A2A receptor mediation [4, 58]. The present study shows that TSP-1 expression in HUVECs is mediated by A2B receptor (Fig. 7). To understand the mechanisms involved in caffeine-induced apoptosis, we evaluated the expressions of Bcl-2 and caspase-3. The results obtained showed that Bcl-2 levels in HUVECs are decreased dose-dependently by caffeine (Fig. 10). On the other hand, caffeine dose-dependently increased the expressions of the proapoptotic forms of caspase-3 (Fig. 9). It has been shown that TSP-1 mediates endothelial cell apoptosis in association with the down-regulation of Bcl-2 and the activation of caspase-3 [28]. In addition, caffeine up-regulated the expression of TSP-1, and it was suggested that caffeine-induced HUVEC apoptosis, at least in part, is association with the up-regulation of TSP-1.
Summarizing, the present study shows that the inhibition of angiogenesis by caffeine is associated with apoptosis related to increased TSP-1 expression due to activation of adenosine A2B receptor.






 XML Download
XML Download