

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Preeclampsia (PE) is a condition unique to pregnancy and characterized by sudden onset of hypertension and proteinurea with maternal dysfunction after 20 weeks of gestation with no previous history of hypertension [1, 2]. Worldwide, PE is estimated to occur in about 5-8% of all pregnancies [3, 4] and it is a major contributor of maternal mortality and morbidity. PE may cause severe maternal disease and premature birth, the perinatal mortality found to be between 1 to 3% [5, 6], being augmentated even three-fold compared to uncomplicated pregnancies [6].
The precise etiopathogenesis of PE still remains under extensive research; while some believe it to be a multifactorial. However the presence of placenta is mainly responsible for the onset and severity of PE [7, 8]. PE is associated with shallow placentation caused by inadequate trophoblast invasion into the maternal spiral artery leads into hypoxic state [9, 10]. This prevents the development of low resistance or high capacitance uteroplacental circulation vital for normal pregnancy. A failure of spiral artery remodelling and foetal vascular development ultimately results in the alteration of villous morphology in PE when compared with normal [11].
In normal gestation as age advances, the terminal villi (TV) are reduced in diameter. The cytotrophoblast in the TV gradually subtle and they contribute their cell mass to growing syncytium [12]. The syncytiotrophoblast is the outer layer of placenta which is in direct contact with maternal blood and is uniquely positioned to alter maternal hemostasis and endothelial function [13]. The syncytium acts as barrier to pathogens and maternal cells and also acts as a secretory and transporting epithelium [14]. An uneven distribution within the syncytiotrophoblast results in clusters of nuclei, the syncytial knots (SKs), which is a focal aggregation or clumping of syncytial nuclei on the outer surface of a tertiary placental villous [15]; SKs are rarely seen in the immature placenta, but gradually increase in number throughout gestation, and at term are present between 10-30% of the TV [16]. The mode of formation and the function of SKs are still far from clear, for they have been variously considered as a degenerative phenomenon [17], an ageing change [18], a syncytial hyperplasia and as a response to trophoblastic ischemia or hypoxia.
The placental villous membrane (syncytioplasm) and the fetal capillaries remain separate but functionally act as a single unit [19], the so called vasculo-syncytial membrane (VSM) [20, 21] which is the only physical barrier between fetal and maternal blood. The most significant properties of the VSM are maintenance of exchange surface area and effective diffusion distance of fetomaternal surfaces [22]. Increased thickness of VSM leads to fetal hypoxia and appears to subject the fetus to considerable risk [23]. Deficient of VSM in hypertensive placental villi can be considered as the failure of trophoblastic differentiation [24]. Almost all the previous placental studies revealed specific rapport between VSM and that of fetal hypoxia. Any morphological or functional alterations of VSM, compiled with other complications may end up with the preeclamptic condition.
The objective of the present study is to compare the morphological and histomorphometrical changes of the normal and PE placentas. The micro anatomical changes of the VSM and SKs were observed, compared and studied in relation to the fetal hypoxia.
Materials and Methods
For the present study, a total of 84 placentas were collected from labor room of Narayana Medical College & General Hospital, Nellore. Forty two placentae were from normotensive pregnancy patients (controls) and the rest forty two from pregnancy complicated by PE. Informed written consent was taken from all the enrolled mothers with proper institutional ethical committee approval. PE were selected under the following criteria's based on the definition of American College of Obstetrics and Gynecologists: 1) Blood pressure (BP) greater than 140/90 mm Hg manifested on two occasions at least 6 hours apart and 2) Proteinurea of 300 mg or greater in 24 hours urine collection or protein concentration of 1 g/l on two occasions of at least 6 hours apart.
Inclusion criteria for controls were normal BP, no proteinurea or any other systemic or endocrine disorder. Exclusion criteria included for both control and PE were no diabetes mellitus, obesity, severe anemia (Hb<6 g%), eclampsia or mothers suffering from any other systemic or endocrine disorder.
Immediately following delivery, the embryonic membranes and umbilical cord were trimmed from placentas. Each placenta was weighed and their diameters were measured. Placental tissue samples were cut on a vertical plane through the full thickness around the umbilical cord insertion and one towards periphery. Tissue samples were placed in 10% formol saline solution for 24-48 hours and embedded in paraffin. The 4 micron thick sections made were stained with hematoxylin and eosin. Microscopic examination was carried out on 1,500 TV randomly on both groups. They were recognized as smallest villi containing capillary loops and completely surrounded by blood. The numbers of capillaries were counted randomly in selected fields per slide using trinocular microscope (CX31, Olympus, Tokyo, Japan) with 40× objective. For VSM thickness with and without SKs, the desired TV were observed using 100× objective. Morphometry of SKs and VSM thickness were studied using Olympus optical micrometers (stage, ocular and reticule). Photographs were taken with Sony DCR W530 digital camera (Tokyo, Japan) fitted on trinocular head.
The data was fed in computer program SPSS ver. 10 for Windows (SPSS Inc., Chicago, IL, USA). The statistical significance of difference between the two groups was evaluated by using Student paired t-test. Data were presented as mean±SD. P-value less than 0.05 was considered statistically significant.
Results
In the present study, 84 (42 each from control and PE) placentas were investigated morphologically and histologically. The mean gestational age was 36.3±3.5 weeks in PE and 38.4±2.7 weeks in control groups (P<0.002).
The mean systolic BP of PE and controls were 144.64±10.55 and 115.2±6.80, respectively while the mean diastolic BP of PE and controls were 97.71±5.5 and 75.47±5.23, respectively. Both systolic and diastolic BP of PE and controls were statistically significant (P<0.001) in this study.
The mean weight of the placenta was found 465.36±81.45 g (range, 250 to 550 g) in controls and 422.38±130.49 g (range, 200 to 700 g) in PE and the difference in weight between two groups were statistically significant (P<0.0001). The neonatal weight and placental weight were significantly reduced in PE than the controls but the feto-placental index did not differ (P>0.9186). No significant difference between the placental thickness and diameter of PE and controls were identified. (Table 1).
In relation to density of SKs, the VSM thickness was twofold increased in PE than the normal but the ratio of VSM thickness without SKs was 1:1.13. The SKs density and diameter was significantly increased in PE than the controls (Table 2). The SKs density was found to be increased in PE than the control and the VSM was also increased in PE than the control.
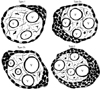
Based on the aggregation around the TV, the SKs in the present study were grouped into type 1, 2a, 2b, and 3 (Fig. 1).
In type 1, SKs were found to accumulate to any one side of the TV leaving back the rest of the circumference of villi allowing free perfusion through VSM (Fig. 2A). This type was commonly found in control and PE (Table 3).
In type 2a, SKs accumulate almost half of the circumference of the TV (Fig. 2B) and only a minimum area related to VSM was left for perfusion. In type 2B, the SKs were collected in two or three small groups surrounding the TV (Fig. 2C). The types 2a and 2b were mostly seen in PE.
In type 3, three fourth of the circumference of TV was covered with SKs (Fig. 2D), thus having the less chances of perfusion, found only in PE.
Gross observations of the placenta in relation to the placental weight and the neonate were found to be statistically significant (P<0.0001) in the PE cases. Histological observation in relation to the VSM and its correlation to the presence or absence of SKs (Fig. 2E, F) were also statistically significant (P<0.0001) with the two times increase in the VSM thickness of the PE placenta.
Discussion
In the present study, VSM thickness was found to be increased in PE and in relation to the SKs it was twofold increased. The SKs density and the diameter were also found to be increased [25]. Both the VSM and SKs studies in PE were statistically significant in the present study. The type 3 SKs classified according to the present study were found only in the cases of PE placenta, in which the VSM thickness was also increased. The aforesaid structural changes in turn causing the functional disturbance of the placenta may result in the hypoxic conditions prevailing in cases of PE [26]. Though type 1 and 2 SKs were also found in PE, the presence of type 3 SKs of the TV along with VSM thickness should be positive basis of fetal hypoxia (Table 3).
Eternally the oxygen in the intervillous space reaches the terminal villus diffused into the fetal capillaries through VSM and finally reaches the fetus via the fetal vascular network [27]. In the present study, the increase in the thickness of the VSM in case of PE reduces the fetoplacental circulation and the release and accumulation of SKs, even makes the condition worst. Increased VSM thickness in PE is associated with maternal uteroplacental vascular pathology resulting in intrauterine distress of fetus [28]. PE is even more complicated frequently by low birth weight and small for gestational age newborns than the normotensive pregnancies [29].
The present study shows a significant difference in the density of SKs and the VSM thickness between the study group (PE) and the control group proposing that the structural alterations of the villous syncytiotrophoblast results in functional impairment of the placenta.
In summary, structural placental changes cause functional placental changes. In this context, morphometry represents an indirect non-invasive study method of placental physiology and pathophysiology. Placental morphology and cellular architecture have the potential factor for oxygen delivery from the mother to the fetus. The placental weight of PE is directly proportional to the development of neonatal birth weight. Next to the placental weight and fetal weight, the gestational age also acts as the most important factor affecting the maternal and perinatal outcome. It is intended in PE that hypoxia injury disrupts the syncytial architecture resulting in the increased density of SKs and VSM thickness that consequently promotes the release of soluble syncytial factors. The increased thickness of VSM causes impaired maintenance of feto-maternal exchange initiating the aponecrosis of syncytiotrophoblast as SKs, subsequently culminating the systemic inflammatory response of the mother. These factors are suggested to pathologically activate the maternal endothelium leading to maternal proteinurea and hypertension, the clinical hallmarks of PE.




 XML Download
XML Download