

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
In the 1950's, the Jackson Laboratory identified two strains of severely obese mice, the ob/ob and the db/db mice that later were found to be precious animal models for the study of obesity and diabetes [1, 2]. It was demonstrated that the ob/ob mice are lacking a lipostatic factor, leptin, a hormone initially found to be secreted by the adipose tissue [3] while the db/db mice were shown to be lacking the corresponding leptin receptor [4]. The brain of both strains of mice is thus lacking a particular signal that triggers the feeling of satiety. Indeed, leptin was shown to reach and interact with areas in the hypothalamus that regulate food intake and energy expenditure. The ob/ob mice lacking the hormone leptin have no sensation of satiety while the db/db lacking of the corresponding receptor has no possibility to convey the signal carried out by the circulating leptin. Both animal strains thus display major food intake problems with related complications ending up being severely obese.
In 1994, Friedman identified and cloned the ob gene in adipose tissue naming the secreted product leptin [3]. Following this, the leptin receptor coded by the db gene was identified and localized in areas of the hypothalamus [4].
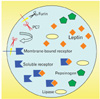
In what concerns the leptin receptor, six isoforms were recognized in rodents, four in humans [4]. Five of them are membrane-bound while one is soluble (three and one in humans). All the receptors have the same extracellular domain and the same affinity for the leptin, but differ in the amino acid sequences and length of their trans-membrane and intracellular domains [5, 6]. The soluble isoform of the receptor, the OB-Re is generated by alternative splicing or by proteolytic cleavage of the membrane-bound isoform [7]. This soluble receptor molecule is released by the leptin secreting cells in the form of a complex with leptin. It is under the form of this leptin-leptin receptor complex that the hormone circulates in the blood. Formation of the complex allows for increasing the half-life of leptin and modulates leptin action on target cells [8].
Leptin is only one of several keys that controls food intake. The feeling of satiety and regulation of food intake is a complex system including several orexigenic and anorexigenic factors that act on the central nervous system and interplay with leptin. Many of them originate from the gastrointestinal track such as cholecystokinin, GLP-1, PYY, ghrelin, insulin while others are adipokines such as adiponectin [9-13].
Adipose Tissue
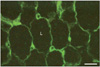
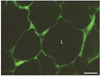
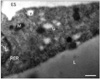
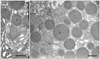
Leptin was first found to be secreted by the adipose tissue [14-18]. Indeed, immunocytochemistry has demonstrated the presence of leptin in the thin cytoplasmic rim surrounding the central lipid droplet of the adipocytes (Fig. 1) and immunoblot confirmed the presence of a peptide of 16 kDa namely leptin in the white adipose tissue homogenate [19]. Electron microscopy has demonstrated that leptin is secreted along the rough endoplasmic reticulum-Golgi-secretory vesicles (Fig. 2) and that this secretion is constitutive rather than regulated [19, 20]. There is no cellular accumulation of a large number of secretory vesicles in the adipocytes, these being discharged as they are formed. When secretion of leptin by the adipose tissue is stimulated, it takes at least 60 minutes for the cells to respond by increasing levels of leptin synthesis and discharge (Fig. 3). Adipocytes also synthesize several of the leptin receptors, the membrane-bound as well as the soluble ones (Figs. 4, 5). Leptin and the soluble receptor are channelled along the same secretory pathway and formed the leptin-leptin receptor complex at the level of the Golgi-secretory vesicle stages before being released. Adipocytes secrete leptin in its complexed form (Fig. 6); leptin is not secreted as a free molecule but always complexed to its receptor [19]. By being complexed to its soluble receptor leptin increases its half life in circulation. We have found that secretion of leptin by the white adipose tissue cannot be the main signal that triggers the feeling of satiety. It is not the factor that controls food intake in the short term. Leptin secreted by the adipose tissue constitutes rather the factor that regulates energy expenditure in steady state conditions [19, 20].
Gastric Mucosa
Leptin gene is expressed by various different tissues including the salivary glands, the placental trophobasts, endocrine glands such as rat pituitary and pancreas and the cardiac and skeletal muscle cells [20]. However, we demonstrated that the gastric mucosa contributes for a large part to the levels of circulating leptin, particularly those registered at time of food intake [21-23].
The gastric mucosa has been shown to secrete large amounts of leptin. Indeed by immunocytochemistry we have demonstrated that endocrine and exocrine cells located in the gastric mucosa are able to secrete leptin either towards the blood circulation or into the gastric juice (Fig. 7) [21]. In what concern the endocrine cells, these are present in the lamina propria of the gastric mucosa (Fig. 7B). These are pure endocrine cells displaying small spherical secretory granules and secrete towards the blood circulation. Double immunolabeling shows that they solely express leptin and its receptor (Fig. 7A) and that, as for the adipocytes, they secrete leptin in its complexed form bound to its soluble receptor. Both molecules are processed along the rough endoplasmic reticulum (RER)-Golgi-granule secretory pathway and leptin get associated to its soluble receptor at the level of the granules. However, the number of these endocrine leptin-secreting cells in the gastric juice is rather limited (Fig. 7A) [20, 21].
In contrast to the few endocrine gastric leptin cells, the large number of chief epithelial cells of the fundic gastric glands expresses and secretes leptin (Fig. 7A). Light and electron microscopy revealed that the epithelial cells of the lower half of the gastric fundus characterized by their secretion of pepsinogen express and secrete large amounts of leptin (Figs. 7, 8) [21]. Working on human gastric tissue [20] we did demonstrate that the leptin-secreting cells are also the ones secreting pepsinogen and lipase (Fig. 8). This is further supported by biochemical and molecular biology data showing that leptin mRNA is present in rat and human gastric mucosa homogenates [24] and that gastric leptin is expressed as monomers and dimers of 16 and 32 kDa respectively and that the pro-leptin is a peptide of 19 kDa.
The chief epithelial cells lining the gastric mucosa secrete leptin along their RER-Golgi-granule secretory pathway (Fig. 9). This secretion has been shown to be regulated, responding very rapidly within minutes to secretory stimuli like food intake and some peptide hormones such as secretin, cholecystokinin and insulin [25-27]. Leptin is synthesised at the level of the rough endoplasmic reticulum, transferred to the Golgi and packaged into the chief cell secretory granules that also contain pepsinogen and lipase [21]. Release of leptin occurs simultaneously with the other granule components into the gastric juice at time of food intake [20].
As for the adipose tissue and the gastric leptin-secreting endocrine cells, leptin is not secreted by the gastric cells as a free molecule but rather complexed to its soluble receptor (Fig. 10A). This soluble isoform of the leptin receptor is generated by the chief cells. The long trans-membrane isoform of the receptor is first synthesized by the chief epithelial cells and packaged into the secretory granule (Fig. 10B). We found it anchored at the level of the membrane of the secretory granules (Figs. 10, 11). It is then converted into the soluble isoform by a cascade of convertases maturation processes including furin and PC7 (Fig. 12). Binding of leptin to its soluble receptor occurs within the secretory granule prior being release into the gastric lumen [17, 20]. The binding of leptin to its soluble receptor is primordial for the integrity of the secreted leptin. Not only binding to its soluble receptor increases the stability and the half-life of the leptin molecule in the blood by several folds allowing it to act for very long time, but in the case of gastric leptin, the binding of leptin to this soluble factor allows leptin to survive the conditions of the gastric juice (Fig. 13) [17, 20]. We have shown that free leptin is immediately degraded upon contact with the gastric juice while the leptin bound to its soluble receptor display a remarkable resistance to the proteolytic conditions of the digestive tract (Fig. 13).
Secretion of leptin by the gastric mucosa is a regulated process triggered by factors quite different from those stimulating adipose tissue leptin secretion. Food intake is a strong stimulus for gastric leptin secretion together with secretin, cholecystokinin, insulin, glucocorticoids, transretinoic acids and pentagastrin [25-27]. In fact, leptin should be submitted to the same stimuli as those triggering secretion of lipase and pepsinogen; however things are somehow more intricate and some agents that stimulate secretion of pepsinogen may have little effect on leptin expression [20].
Gastric Versus Adipose Tissue Leptons
Many differences can be found between adipose tissue and gastric leptins. While the molecules are identical, their secretion by both tissues involves the same leptin-leptin receptor complexes and they interact with the same trans-membrane receptor, major differences exist in the process of secretion and in the factors triggering their secretion. As mentioned above adipose tissue secretes leptin continuously along a constitutive secretory pathway while the gastric cells act along a regulated secretory pathway. Upon stimulation it takes about 60 minutes to register an increase in leptin secretion by the adipocytes. On the other hand, the gastric mucosa reacts immediately upon stimulation of secretion. Thus, while gastric leptin may act rapidly and transiently to certain stimuli like food intake to trigger in the short term the feeling of satiety, adipose tissue leptin may in turn act on the long-term regulating energy expenditure. In addition, gastric leptin exerts a regulatory function on the digestive tract [28, 29]. Intestinal epithelial cells express leptin receptors on both their luminal brush border membrane as well as on their baso-lateral membrane (Fig. 14) [21, 28, 30, 31]. Thus, leptin either originating from the gastric juice or from the blood circulation can act on the intestinal cells to regulate digestion of nutrients.
Gastric Leptin in the Intestinal Tract
We have demonstrated that upon surviving the gastric juice conditions, gastric leptin is vehiculated towards the duodenal lumen. Intestinal enterocytes express trans-membrane leptin receptors at the level of their microvilli apical membrane as well as at the level of the basolateral membrane (Fig. 14). Thus, gastric leptin upon reaching the duodenal lumen interacts with its membrane receptors and acts on the activities of the intestinal absorptive cells. On the other hand, circulating leptin acts on the baso-lateral side of the same cells again influencing intestinal activities. The luminal leptin exerts its action in a paracrine fashion while the circulating one acts rather as an endocrine hormone. Several aspects of the intestinal physiology are influenced by leptin. Among many, leptin influences nutrients absorption, mucus secretion, intestinal motility and inflammation. The transport, processing and delivery of nutrients to the systemic circulation are directly or indirectly modulated by endocrine and exocrine factors originating from the gastro-enteric wall such as leptin [32]. Binding of leptin to its luminal enterocyte receptors activates several intracellular pathways such as the protein kinase C (PKC) [33] and MAPkinase-dependant pathways [34] as well as the STATs pathways [28]. Leptin is able to decrease the sodium-glucose-transporter 1 inhibiting glucose uptake in a PKC-dependant manner not affecting the D-galactose uptake [33]. In what concerns amino acids uptake by the intestinal enterocytes, leptin displays a positive effect increasing the density of the transporter pepT1 [35]. On the other hand, leptin decreases release of triglycerides into the baso-lateral medium as well as that of apolipoprotein B-100 and B-48 as well as the chylomicrons and low density lipoproteins. Leptin increases synthesis of apolipoproteins A-1 and Apo A-V and Apo E while the intestinal absorption of cholesterol is decreased [36, 37].
Transcytosis of Gastric Leptin across the Intestinal Enterocytes
When we followed variations in circulating levels of leptin, we can readily detect that plasma leptin rises significantly and rapidly within 15 minutes after the onset of food intake or upon cholecystokinin stimulation [22]. Leptin from white adipose tissue cannot account for such a raise of circulating leptin since as we mentioned above, it takes over 60 minutes, for adipose tissue to respond to any secretory stimuli [20]. The hypothesis put forward was that the sudden increase in circulating leptin seen upon food intake, may originate from the postprandial leptin secretion occurring into the gastric juice. This would require that the gastric leptin reaches the in testinal lumen and would have the ability to cross the intestinal mucosa and reach the blood circulation without being altered. We have previously demonstrated that several large proteins and peptides present in the duodenal lumen do cross the intestinal barrier and reach the circulation retaining their molecular integrity and biological activity [31, 38-40]. The fact that gastric leptin does reach the blood circulation by crossing the intestinal barrier was demonstrated by monitoring the rise of plasma leptin levels upon cholinergic stimulation of gastric leptin secretion and maintaining or not the pyloric sphincter open (Fig. 15) [31]. Indeed, closing the sphincter prevented this rise in circulating leptin, thus indicating that gastric leptin does contribute to plasma levels variations at time of food intake (Fig. 15). A second experiment that came in support of our hypothesis was carried out by using leptin tagged with a tracer, such as fluorescein isothiocyanate (FITC) [31]. Upon introducing leptin-FITC in the rat duodenal lumen, the complex appeared in the blood in a time-dependant and saturable manner (Fig. 15). Light and electron microscopy supported by immunocytochemistry revealed the pathway taken by the leptin-FITC to cross the intestinal barrier (Figs. 16, 17). Upon binding to the luminal trans-membrane luminal receptor, leptin is internalized by the intestinal cells through endocytotic vesicles. Leptin is then channelled to the Golgi apparatus remaining only in the most trans-Golgi cisternae. Leptin is then packaged in secretory vesicles and transferred to the baso-lateral membrane where it is released into the interstitial space (Fig. 17). Leptin diffuses in the interstitial space and reaches the blood circulation across the capillary endothelial cells (Fig. 17). Leptin was not detected at the level of the epithelial intercellular junctions, validating the transcytotic transport across the intestinal epithelial cells rather than a para-cellular pathway. Along this trans-cellular pathway, different processes were revealed. We have demonstrated that in spite of the fact that free leptin was introduced into the lumen of the duodenum a leptin-soluble leptin receptor complex was released by the duodenal enterocytes. The exogenous leptin acquires the soluble receptor and binds to it to form a complex at the level of the trans-Golgi cisternae. We have demonstrated that intestinal enterocytes do synthesize the leptin receptor. They express the long-isoform of the receptor at the level of the plasma membrane, the apical as well as the baso-lateral ones. They further convert the long isoform into it the soluble one that binds leptin in the Golgi before being released as a complex (Fig. 18) [31]. The transcytotic transport of leptin from the intestinal lumen to the baso-lateral interstitial space differs from classical transcytotic phenomenon by the fact that the transported molecule transits through the Golgi. This transit through the Golgi apparatus is not a common process in transcytosis but is needed in the case of leptin for the leptin-leptin receptor complex to be formed prior release. Such particular transcytotic transport of leptin across the enterocyte was confirmed by in vitro studies using the Caco-2 human intestinal cell line which is a quite appropriate in vitro model for the study of the intestinal mucosa [41]. Caco-2 cells were demonstrated to express trans-membrane leptin receptors at both their apical and baso-lateral membranes. Similarly to what was found in vivo, free leptin introduced on the apical chamber of the culture system was internalized by the Caco-2 cells at the level of their apical plasma membrane, transferred to the basolateral membrane and secreted into the basal chamber of the culture. This transport was found to be time and temperature-dependent and is saturable, suggesting that it is receptor-mediated (Fig. 18). This receptor-mediated transport was demonstrated by molecular biology knockdown experiments revealing that the long trans-membrane leptin receptor isoform is the one involved in the endocytosis and transcytosis of leptin (Fig. 18). As observed in vivo, inter nalised leptin from the luminal membrane is channelled to the Golgi apparatus prior to be secreted on the basal side of the cell. Drug disruption of the Golgi apparatus significantly decreased the transport of leptin. Also, as found in vivo, the free leptin introduced in the apical chamber of the culture, was secreted in the basal chamber bound to its soluble receptor. Caco-2 cells synthesize the leptin receptor and form the leptin-leptin receptor complex at the level of the Golgi. The complex is secreted by the Caco-2 cells in the basal chamber [41].
Conclusion
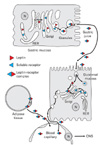
Fig. 19 summarizes the molecular and cell biology processes leading to the secretion of leptin. On the one hand, the adipocyte synthesises both the leptin and the soluble isoform of the leptin receptor, generating the leptin-leptin receptor complex along the secretory pathway. This complex is released by the cell through a constitutive secretory process. Secretion takes place towards the interstitial space; the complex diffuses and reaches the blood circulation across the blood capillary endothelial cells. On the other hand, the gastric chief cell also synthesizes both the leptin and the leptin receptor. These are channelled along the regulated RER-Golgi-granules secretory pathway. At the level of the secretory granule, the transmembrane leptin receptor is converted into its soluble isoform binding the leptin molecules present in the same secretory granule to form the leptin-leptin complex. In contrast to the adipose tissue, the gastric chief cell accumulates the secretory granules in its cytoplasm till secretory stimuli trigger their discharge. The granule content is discharged into the gastric juice. The formation of the complex protects the leptin molecules from being degraded rapidly in the gastric juice. Leptin is then channelled to the duodenal lumen where it interacts with the transmembrane leptin receptor expressed on the apical membrane of the duodenal enterocyte. Upon binding this receptor, leptin is internalized by the duodenal enterocyte through its endosomal compartment. In the early intracellular stages, leptin gets separated from its receptor and continues its way to the trans-Golgi cisternae. In the Golgi cisternae, leptin binds a newly synthesized molecule of its soluble receptor to form again a new leptin-leptin receptor complex. The complex is then packaged in small secretory vesicles and discharged on the baso-lateral membrane of the enterocyte into the duodenal interstitial space. The complex diffuses and reaches the circulation crossing the blood capillary endothelial cells.
Both the adipose tissue and gastric leptins reach the hypothalamic targets cells via the blood circulation to act either for the long term regulation of energy expenditure (the adipose tissue leptin), or for the regulation in the very short term of food intake (the gastric leptin).
Leptin and obesity
Obesity has become a major global health problem. Although it may result from defective genes that trigger metabolic problems, in its majority, it results from an unhealthy lifestyle in term of food intake and energy expenditure. Few individuals bear homozygous nonsense mutation of leptin or of the leptin receptor. Such mutations induce a lack in leptin-mediated satiety signal in the brain leading to hyperplasia and morbid obesity. Such situations are encountered in mice with the two mouse models, the ob/ob mice that lack the leptin and the db/db mice lacking of the functional leptin receptor. In the case of leptin deficiency, i.v. injections of leptin may help in reducing food intake, increase energy expenditure and eventually in losing body weight. This has been successfully achieved in ob/ob mice [42, 43]. In the human, such total lack of leptin is quite rare and thus i.v. treatment of obese patients with leptin alone does not reach such successful results as in the mutated mice. Additional problems with obesity concern the various pathologies that are associated. Indeed severe obesity leads to the development of numerous other pathologies related to diabetes and coronary diseases. Novel approaches and additional knowledge on leptin secretion, leptin handling by tissues, leptin receptor expression and the most conspicuous roles played by the soluble isoform of the leptin receptor should contribute significantly to our understanding of the physiology of food intake and energy expenditure. As it stands now, levels of circulating leptin result from the contribution of two major organs, the white adipose tissue and the gastric mucosa. In both cases the end results concern the complex leptin-leptin receptor that is quite stable, remaining for long periods in circulation and allowing for optimal interactions with hypothalamic and other target cells.










 XML Download
XML Download