

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Retinal degeneration (RD) is a general cause of blindness. It is a clinically and genetically highly heterogeneous disorder affecting many people the world wide [1]. This hereditary disease is characterized by the progressive degeneration of photoreceptor cells in the retina. It first influences rods and later results in the loss of cones. Eventually, cone degeneration affects the central vision, leading to complete blindness [2, 3]. In order to approach available clinical treatments for RD, studying its pathophysiology and evaluating the effects of new therapeutic agents are essential. However, an important issue in this field is the need for reliable and stable animal model for RD research.
To date, there have been many hereditary animal models for RDs [2, 4-6]. In addition to hereditary animal models, there are various chemically induced RD models [7] which are easier to produce and in which it is easier to modulate degrees of change. One of the chemicals, N-methyl-N-nitrosourea (MNU), is a potent retinotoxin which leads to apoptotic cell death of photoreceptor cells [8-15]. Although several reports have shown that MNU was very effective chemical tool to induce RD in animal models, there are very few studies to examine morphological changes and correlation with functional responses of RD induced by MNU [13, 16]. Thus, this evaluated a rat model in which RD was induced by MNU. We examined morphological changes of the retina induced by MNU and evaluated retinal function by means of electroretinogram (ERG) according to changes in MNU dose. We determined the appropriate dosage of MNU for the study of the RD. In addition, we examined morphological and functional changes in the degenerating retina induced by MNU in the animal model over time. Our results suggest that this RD rat model may be a useful model to study its pathophysiology and to evaluate the effects of new therapeutic agents before clinical trials.
Materials and Methods
Animals
One hundred and seven 10-week-old male albino Sprague-Dawley (SD) rats were used. All animals were treated according to the regulations of the Catholic Ethics Committee of the Catholic University of Korea, Seoul, which conform to the National Institute of Health (NIH) guidelines for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996.
RD model
Fifty four SD rats received a single intraperitoneal injection of 1% MNU in freshly prepared saline (40, 50 and 60 mg/kg; Sigma-Aldrich, St. Louis, MO, USA). One week after MNU treatment, rats were sacrificed with 8% chloral hydrate (0.5 ml/kg) after ERG for visual function tests. Thirty two SD rats injected with 50 mg/kg MNU were also sacrificed 2 weeks and 4 weeks after MNU treatment. Twenty one control animals (12 for dose-dependent effects of MNU and 9 for time-dependent effects of MNU) were treated with normal saline.
ERG
All rats were dark adapted for 1 hour before ERG experiments and were prepared for recording under dim red light (λ>600 nm). Anesthesia was initially induced with an intraperitoneal injection of 8% chloral hydrate (0.5 ml/kg). Supplemental anesthesia was provided at approximately 1 hour intervals (8% chloral hydrate 0.2 ml/kg). Animals were lightly secured to a stage to ensure a stable position for recording the ERG. The top of the stage was fixed to the position where the animal eye faced the flash light at a 20-cm distance. Depth of anesthesia was sensitively monitored online by inspection of the ERG signal baseline. The cornea was protected with hydroxypropyl methylcellulose gel. A gold ring contact electrode was placed on the cornea. A reference electrode and a ground electrode were placed subcutaneously in the ear and in the tail, respectively. Stimuli were brief white flashes (xenon arc discharge) delivered via a Ganzfeld integrating sphere (Model UTAS-3000, LKC Technologies, Gaithersburg, MD, USA). Stimulus intensities were measured using a calibrated photometer with a scotopic luminosity filter in place. Because the rat retina is particularly rod-dominant, we fixed all flash stimuli at scotopic intensities, which better reflects the retinal function in this animal. Scotopic ERGs obtained for all intensities above 0.9 log (cd s) m-2 were recorded as single flash responses [16, 17]. Each record was an average of 6 responses obtained with a 2 second interstimulus interval. Signals were recorded with band-pass filters of 1-3 Hz. The amplitude of the a-wave was measured from the baseline to a-wave trough, and b-wave was measured from the maximum a-wave trough to the maximum b-wave peak.
Statistics
Statistical analysis was conducted by ANOVA and Student's t-test to compare ERG differences between control and MNU-treated groups. Data are represented as mean±standard deviation. The differences were considered statistically significant at P<0.05.
Tissue preservation
The anterior segments of the eyeball were removed. Eyecups were fixed by immersion in 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) for 3 hours. They were rinsed in PB, dehydrated in a graded series of ethanol and embedded in wax (polyethylene glycol 400 disterate, Polysciences, Warrington, PA, USA). Some 5-µm vertical sections of the eyecup were made, dewaxed and rinsed in PB. They were stained with hematoxylin and eosin (H&E).
Immunohistochemistry
Five micrometer vertical sections were used. They were incubated in 10% normal donkey serum in PB for 1 hour at room temperature, to block any nonspecific binding sites. They were then incubated with a monoclonal antibody against glial fibrillary acidic protein (GFAP; 1 : 1,000, Chemicon, Temecula, CA, USA) in PB for 1 day at 4℃. The sections were washed in PB for 30 minutes (3×10 minutes) and incubated in the presence of Cy3-conjugated donkey anti-mouse IgG (1 : 1000, Jackson ImmunoResearch, West Grove, PA, USA) for 2 hours. After thoroughly washing with PB, the wax embedded tissues were mounted with Kaiser's glycerol gelatin.
Results
Dose-dependent RD induced by MNU injection
Functional assessment by ERG
ERG recordings were taken to evaluate retinal function of a RD model induced with MNU according to changes in MNU dosage. All animals treated with MNU showed reductions in both a- and b-wave amplitudes 1 week after MNU injection (Fig. 1). When 40 mg/kg MNU were applied, both ERG responses were reduced compared to normal (a-wave, 20.6±3.0 µV vs. 22.1±2.7 µV; b-wave, 55.9±4.4 µV vs. 62.8±7.8 µV), but differences were statistically insignificant (P>0.05). However, when 50 and 60 mg/kg MNU were applied, both ERG responses were significantly reduced by 26% and 44% in a-wave and 35% and 42% in b-wave (P<0.05), respectively, compared to control rats (a-wave, 16.3±2.0 µV and 12.4±1.6 µV vs. 22.1±2.7 µV; b-wave, 40.9±4.3 µV and 36.2±8.2 µV vs. 62±7.8 µV). Fig. 1C shows representative waveforms of ERG responses in control and MNU-treated eyes with different doses.
Histological aspect by H&E staining
Histopathological changes were evaluated in H&E stained retinal vertical sections of the control and MNU-treated rats 1 week after injection (Fig. 2). The layer number of photoreceptors in the outer nuclear layer (ONL) slightly decreased in 40 mg/kg MNU-treated rat retinas (Fig. 2B) compared to control rats (14-17 layers vs. 12-14 layers). In addition, the arrangement of photoreceptors in the ONL was slightly distorted and the retina appeared to be edematous, especially the inner plexiform layer (IPL) (Fig. 2B). In 50 mg/kg MNU-treated rat retinas (Fig. 2C), 10-12 layers of photoreceptors remained, the edematous appearance in the retina continued, and the distortion of the inner nuclear layer (INL) was apparent. In 60 mg/kg MNU-treated rat retinas, numerous photoreceptors and bipolar and amacrine cells were degenerated and thus, thicknesses of the OPL and the IPL were markedly decreased (Fig. 2D). Taken together, our functional and histological findings suggest that degenerative changes in the retina treated with MNU are dose-dependent.
Retinal injury evaluation by immunohistochemistry with anti-GFAP
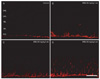
By immunohistochemistry with anti-GFAP, a reliable marker for profiling neurotoxicity [18, 19], we evaluated the degree of retinal injury according to the MNU injection in different doses. In control retinas, GFAP was expressed within end feet portion of Müller cells and astrocytes located in the ganglion cell layer (GCL) (Fig. 3A). Similar to the finding in control retinas, GFAP immunoreactivity was found mainly in the GCL, but GFAP-immunoreactive profiles appeared to become thicker in 40 mg/kg MNU-treated retinas (Fig. 3B). The expression of GFAP increased more in 50 mg/kg MNU-treated retinas than in 40 mg/kg MNU-treated retinas and thus, some labeled radial processes of Müller cells running in the IPL were observed (Fig. 3C). In 60 mg/kg MNU-treated retinas, some GFAP-immunoreactive Müller cell processes extended to the distal region of the retina passing through the INL (Fig. 3D). These results demonstrate that RD in the rat model with MNU injection is dose-dependent.
Time-dependent RD induced by MNU injection
Functional assessment by ERG
The initial part of our study showed dose-dependent RD effects of MNU. Next, we wanted to know whether RD by MNU treatment is time-dependent. Thus, we examined changes in the function and histology over time in 50 mg/kg MNU-treated animals, which demonstrated significant changes in ERG responses, apparent decreases of photoreceptor layers and increases in GFAP expression.
Fig. 4 shows MNU effects on ERG responses over time. Amplitudes of both a- and b-waves were reduced at 1 week by 37 and 29%, respectively, compared to those in control animals (a-wave, 24.2±3.8 µV vs. 38.6±4.3 µV; b-wave, 68.4±11.1 µV vs. 95.9±6.0 µV) (Fig. 1). At 2 and 4 weeks, both ERG responses became progressively reduced by 47% and 67% in a-waves and 45% and 75% in b-waves, respectively (a-wave, 20.5±5.2 µV and 12.8±5.8 µV; b-wave, 52.6±12.1 µV and 33.4±6.5 µV), compared to those in control animals. Fig. 4C shows representative waveforms of ERG responses in control and MNU-treated eyes at different time points.
Histological aspect by H&E staining
As shown in Fig. 2B, in retinal vertical sections taken from a retina 1 week after 50 mg/kg MNU injection, loss of 2-3 photoreceptor layers, disruption of the arrangement of the INL, and retinal edema were observed (Fig. 5B). These degenerative changes gradually progressed: the number of photoreceptor cell layers was 8-10 two weeks after MNU injection (Fig. 5C) and was only 5-7 four weeks after. Thus, the final retinal thickness was about 56% of that of the control retina (Fig. 5D). These functional and histological findings suggest that degenerative changes in the retina treated with MNU are time-dependent.
Retinal injury evaluation by immunohistochemistry with anti-GFAP
In retinas after the 50 mg/kg MNU injection, GFAP expression increased in a time-dependent manner (Fig. 6). Over time, the number of GFAP-labeled radial processes of Müller cells increased and the length of GFAP-immunoreactive processes was extended and thus, GFAP-immunoreactive Müller cell profiles were frequently observed in the INL (Fig. 6D). These results demonstrate that RD in the rat model with MNU injection is time-dependent.
Discussion
Recently, there has been an increasing need for a well-documented animal model for RD which can be used to study of RD as a heterogeneous group of human disease causing blindness. In the present study, we aimed to assess whether the rat model with MNU injection is of value as a RD animal model. We examined changes in function and histology in rat retinas ongoing RD evoked by MNU injection with different dosages and time points. Upon MNU injection of 40, 50 and 60 mg/kg, ERG responses showed reduction in amplitudes of both a- and b-waves compared to control responses. At 1 week, 2 weeks and 4 weeks after MNU injection (50 mg/kg), amplitudes of both a- and b-waves also decreased. Consistent with these functional changes, histopathologic findings showed MNU dose- and time-dependent RD and expression of GFAP, a marker for retinal injury, increased. This correlation among the results indicates that the rat RD model with MNU injection is of great value for the study of RD in human.
In humans, a representative RD disease is age-related macular degeneration (AMD) which is the leading cause of blindness in the elderly in developed countries [20]. The cause of AMD is complex and many risk factors have been suggested, including age, family history (genetics), diet, smoking, and other environmental risk factors. The cellular components involved in the disease process are photoreceptor cells and retinal pigment epithelium (RPE), which gives metabolic support to the photoreceptor cells. In addition, the Bruch membrane, a complex of collagen and elastic layers interposed between the RPE and choroid where a capillary bed provides the metabolic needs of the outer retina, is another key component affected [21]. To date, numerous inherited and chemically-induced animal models have been introduced. In inherited animal models, rd1 mouse mutated in the β-subunit of rod cGMP phosphodiesterase (Pdebrd1) [4, 6, 22] and rds mouse mutated in the retinal degeneration slow-peripherin (Prph2Rd2) [5] have been well-documented. Both strains have shown similar pathologic events found in human AMD, affecting photoreceptors and RPE. However, inherited animal models have early onset of RD, while human AMD generally has a later onset. In addition, the development of drusen, subretinal deposits composed of a large number of different components including acute phase proteins, lipid, b-amyloid, and complement proteins, is rarely seen in animal models, even in aged genetically modified mice [23].
Chemically-induced animal models have also been used to gain insights into human AMD. There have been many studies to introduce several chemicals as potent retinal neurotoxins to induce RD. One of them, sodium iodate (NaIO3), is a stable retinotoxin that has been used as a RD animal model [17, 24]. However, two other study groups demonstrated a mismatch between visual function and histopathology of the retina [25, 26]. Additionally, in our preliminary test, NaIO3 was not likely to reduce ERG responses significantly and or show apparent histopathologic findings (data not shown). However, the present study clearly shows that MNU reliably and reproducibly can cause RD. We also demonstrated that RD in this model shows correlation between visual function and histopathology of the retina, and MNU dose- and time-dependency.
MNU used in this study has been widely used to develop a RD animal model for the evaluation of the effects of new therapeutic agents since Smith et al. [27] reported that MNU provokes a progressive RD. However, research groups have made RD animal models with MNU in a wide range of dosage,s from 40 mg/kg to 60 mg/kg [9, 10, 13, 16], indicating the absence of established protocol for MNU treatment. Most studies have focused on the investigation of whether short-term supplementation of certain agents can overcome more severe retinal damage caused by higher dose of MNU (>60 mg/kg) [13, 16, 28, 29]. To evaluate the effects of new therapeutic agents before clinical trials, long-term administration is prerequisite for toxicity tests. Thus, we focused on the long-term progress RD induced by appropriate doses of MNU in an animal model. We found that RD induced by MNU is dose-dependent. However, administration of 40 mg/kg MNU did not show a significant reduction in ERG responses 1 week after MNU injection. Both 50 mg/kg and 60 mg/kg MNU injections showed significant decreases in ERG responses. In the case of 60 mg/kg MNU injection, severe RD occurred and only about half of photoreceptor cells remained 1 week after MNU injection. Thus, we concluded that a dose of 50 mg/kg is appropriate for long-term observation in a RD animal model with MNU injection. Our conclusion was corroborated by the findings that functional and histological findings showed time-dependent RD to 4 weeks when 50 mg/kg MNU was applied.
ERG represents the total effects of various retinal neurons, pigment epithelium, and Müller cells [30, 31]. In flash of light stimulation, a- and b-waves are most often observed. The a-wave is a negative component and reflects function of photoreceptors. The b-wave is a corneal positive component and reflects ON bipolar cells and Müller cells. Because MNU induces selective photoreceptor cell death, we expected a marked reduction in a-waves and a relatively small reduction in b-waves. However, similar reductions in both a- and b-waves were detected. These findings may be due to the relatively late observation time points. That is, we observed dose- and time-dependent effects of MNU 1 week after injection and afterwards. Based on previous reports that photoreceptor degeneration has been observed 24 hours after MNU injection [9, 10, 14], our ERG results showing similar reductions in both a- and b-waves indicate that bipolar cells might be suffering secondary degeneration at the time points when we examined in the RD model used in this study.
To evaluate the degree of retinal injury according to MNU injection in different doses, we observed GFAP expression by immunohistochemistry. GFAP is a reliable marker for profiling neurotoxicity [18, 19]. As expected, GFAP expression correlated with the degree of RD that is MNU dose- and time-dependent. These results indicate that GFAP is a good marker for the evaluation of RD.
In conclusion, a RD rat model with MNU injection shows RD in dose- and time-dependent manners. In this study, an application of 50 mg/kg MNU may be the best protocol for studying RD progression, mechanisms, and evaluation of long-term effects of new therapeutic agents.





 XML Download
XML Download