

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Exposure to ethanol during gestation induces abnormalities of structure and function on neuronal maturation, resulting in fetal alcohol syndrome (FAS) in the brain [1-4]. FAS is a specific, recognizable pattern of malformation, resulting from exposure of the human fetus to ethanol and leading to developmental disorders, including central nervous system (CNS) dysfunction, neurobehavioral disruption, neurological deficits, and epilepsy [5, 6]. Among the major neuropathological changes of FAS, size of the hippocampus and other brain regions is reduced [2, 7], but the mechanism by which ethanol induces this damage in the prenatal hippocampus is not fully understood [8].
γ-aminobutyric acid (GABA), the principal inhibitory neurotransmitter, has been heavily investigated for a possible role in ethanol-mediated neuropathology. Previous work focused on the GABAAR chloride channel [9], which has been demonstrated to contribute to FAS [10]. The GABAB system, the predominant inhibitory metabotropic receptor in the mammalian CNS, has been less well characterized due to the limited number of pharmacologic agents that can interact with GABABR [11], which mediates slow inhibitory GABA synaptic neurotransmission through association with G proteins and through cAMP-dependent signaling [12, 13]. Some studies have indicated that the GABABR agonist baclofen can reduce ethanol intake, although this may depend on the dose administered [14-17]. In addition, the GABABR antagonist phaclofen attenuates the coordinating and locomotor-activating effects produced by low doses of ethanol. Moreover, phaclofen reduces some of the behavioral alterations produced by anesthetic concentrations of ethanol [18]. RNA interference (RNAi) against the GABAB1R or treatment with the GABAB antagonist CGP 54626 reduces ethanol's impairment of behavior, possibly implying a role for GABABR in mediating ethanol's neurological effects [19]. Furthermore, the cAMP-signaling pathway is a major target of ethanol in the CNS [20]. It has been previously reported that the activation of protein kinase A (PKA) is likely to be responsible for cAMP response element binding protein (CREB) phosphorylation, and in the hippocampus, stimulation of adenylyl cyclase either by Ca2+/calmodulin kinase (CaMKII) or by receptor-coupled G proteins increases intracellular concentrations of cAMP. Thus, the GABAB system may transduce ethanol's effects [11].
In the present study, we used an agonist and an antagonist of GABABR, as well as siRNA treatment, to study the modulation of GABAB1R, PKA-α, CaMKII, and p-CREB expression in cultivated hippocampal neurons in vitro under ethanol treatment (ET).
Materials and Methods
Animal treatment
Female (n=30) Sparague-Dawley rats (250 g, Gyeongsang National University Neurobiology Laboratory, Jinju, Korea) were housed in a temperature-controlled environment with lights on from 6:00-20:00 h and food ad libitum. Pregnant rats were killed by ethyl ether anesthesia at gestational day (GD) 17.5 of fetal development.
Primary hippocampal cell culture
Primary hippocampal cultures were prepared from brains of GD 17.5 fetuses. Hippocampal tissues were treated with 0.25% trypsin-EDTA for 20 minutes and dissociated by mechanical trituration in ice-cold calcium- and magnesium-free Hank's balanced salt solution. After pelleting by centrifugation, cells were plated (1×106 cells/ml) in cell culture plates pre-coated with poly-lysine (0.02 g/L) and chamber slides. The culture medium consisted of Dulbecco's modified Eagle medium (DMEM) with 10% heat-inactivated fetal bovine serum, 1 mM pyruvate, 4.2 mM sodium bicarbonate, 20 mM HEPES, 0.3 g/L bovine serum albumin, 50 U/ml penicillin, and 50 mg/L streptomycin. Cultures were maintained at 37℃ in a humidified atmosphere with 5% CO2. Neuroglial cell proliferation was inhibited by treatment with growth medium containing 10 µM cytosine β-D-arabino furanoside (Sigma, St. Louis, MO, USA) for 12 hours.
siRNA transfection and drug treatment
GABAB1R cDNA was removed from pCI vector by XbaI and EcoRI digestion (provided by Novatis Pharma, Basel, Switzerland). The primer which contained T7 promoter region (forward: 5'-CGGTAATACGACTCACTATAGGGAG ACGCTACCATCCAACAGACCA-3', reverse: 5'-GCGTAA TACGACTCACTATAGGGAGATCCTGTGAGCTCATGTT GGAA-3') was used for amplification of cDNA of GABAB1R. Then dsRNAs were prepared with the MEGAscript® RNAi kit (Ambion, Austin, TX, USA) by incubation at 37℃ for 4 hours. RNAi was performed by using a ShortCut RNAi Kit (New England Biolabs, Buckinghamshire, UK). For GABAB1R (420 bp) from 1,096 bp to 1,516 bp (accession no. NM 031028) with the highest silent activity was selected. One day before transfection, the medium was changed to DMEM without antibiotics and serum. Liposome solution (DMEM containing Lipofectamine2000™, Invitrogen, Carlsbad, CA, USA) and equal volume of dsRNA (21 bp as shortcut dsRNA) solution were incubated for 5 minutes at room temperature respectively, then combined together and incubated for 20 minutes. The mixture was added to the culture medium in each plate to reach a final concentration of 20 nM. After 48 hours of transfection, both cells with and without RNAi transfection transfer to the normal media as control, media contained 100 mM ethanol, 100 mM ethanol and 50 µM baclofen, 100 mM ethanol and 100 µM phaclofen, 100 mM ethanol and 50 µM baclofen plus 100 µM phaclofen. After 20 minutes of drug treatment, the cells were harvested. And the transfection efficiency was confirmed by using a BLOCK-iT™ transfection kit (Invitrogen) containing a fluorescent oligonucleotide (100 mM) for siRNA transfection after 2 days.
Reverse transcription polymerase chain reaction (RT-PCR)
RT-PCR analysis was performed to identify cDNAs transcribed in transfected or mock-transfected cultures. Total RNA was isolated with TRIzol Reagent (Life Technologies, Rockville, MD, USA). First strand cDNA was transcribed from 2 µg RNA using oligo (dT)15 and M-MLV reverse transcriptase (Promega, Madison, WI, USA) according to manufacturer's instructions. A total of 4 µl cDNA were used for PCR amplification with 1 µl Taq DNA polymerase. Thermal cycling was performed under the following conditions: 94℃ for 5 minutes; 30 cycles at 94℃, 68℃, and 72℃ for 1 minute each; final extension at 72℃ for 5 minutes. A negative control PCR for β-actin was performed at 58℃ for 25 cycles. PCR product was run out on a 1% agarose gel containing ethidium bromide and viewed under UV light. The primers used were the following:
GABAB1R forward primer: 5'-AATTGAATTCCGCTACCATCCAACAGACCA-3';
GABAB1R reverse primer: 5'-AATTAAGCTTTCCTGTGACGTCATGTTGGAA-3'
Western blotting
Drug-treated hippocampal cells were homogenized in cell lysis buffer (Cell Signaling no. 9803, Danvers, MA, USA) for protein extraction. After two rounds of ultra-centrifugation at 12,000 rpm for 10 minutes (4℃), supernatant protein concentration was quantified using the Bio-Rad protein assay kit. Thirty µg soluble fraction were separated on duplicate 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels (30% acrylamide, 1% Bis, 1 M Tris, 20% SDS, 10% APS, TEMED). Proteins were transferred to polyvinylidene fluoride membrane at 90 V for 1 hour (in electrotransfer buffer with 48 mM Tris, 39 mM glycine, 20% MeOH, and 0.037% SDS). Membranes were treated with blocking solution (Tris-buffered saline containing 0.1% [v/v] Tween-20 and 6% [w/v] non-fat dry milk) to reduce non-specific binding. Immunostaining was carried out using a guinea-pig derived anti-rat GABAB1R (1 : 500, 24 hours, 4℃, Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-rat PKA-α (1 : 500, Santa Cruz Biotechnology), rabbit anti-rat CaMKII (1 : 500, Cell Signaling) or rabbit anti-rat p-CREB (1 : 500, Cell Signaling). Anti-Actin antibody (1 : 1,000, Sigma-Aldrich, Jerusalem, Israel) was taken as control to confirm uniform loading. Antigen was detected by enhanced chemiluminescence (Western blotting detection reagents, Amersham Pharmacia Biotech, Piscataway, NJ, USA) according to manufacturer's instructions. In some cases, the blots were stripped and reprobed. Briefly, after film exposure, washed membrane four times for 5 minutes each in TBST, then agitated gently the membrane in the antibody stripping solution (ReBlot Plus Strong Antibody stripping solution, Millipore, Temecula, CA, USA) at room temperature for 15 minutes. After that, again washed the membrane four times for minutes each in TBST, then reprobed with a rabbit antiactin (1 : 1,000, Sigma-Aldrich). Western blots were analyzed by densitometry using the computer-based Sigma Gel system (SPSS Inc., Chicago, IL, USA). Density values were expressed as mean±SEM. A Student's t-test was performed to determine significant differences among groups. In each case, significance was accepted to be P<0.05.
Immunofluorescence assays
Immunofluorescence was performed on GD 17.5 primary hippocampal cell cultures grown in vitro on poly-L-lysine coated round glass coverslips. Cultures were fixed for 5 minutes with 4% paraformaldehyde in phosphate buffered saline (PBS), permeabilized in 0.3% Triton X-100 in PBS for 5 minutes, and rinsed twice in PBS. The cells were preincubated with 10% bovine seum albumin in PBS for 1 hour at 37℃ before exposure to primary antibodies overnight at 4℃. Immunostaining was carried out using guinea-pig derived anti-rat GABAB1R antibody (1 : 500, 24 hours, 4℃, Santa Cruz Biotechnology), rabbit anti-rat PKA-α antibody (1 : 500, Santa Cruz Biotechnology), or rabbit anti-rat anti-p-CREB antibody (1 : 500, Cell Signaling). Following incubation, cells were washed and secondary antibodies applied as follows: anti-rabbit IgG-TRITC and -FITC conjugated antibodies, 1 : 100; anti-guinea pig IgG-FITC conjugate, 1 : 150 (both from DAKOCytomation, Glostrup, Denmark). For co-staining, incubations were performed in parallel. Glass cover-slips were affixed to slides with mounting medium. Imaging was performed with a fluorescence microscope (Zeiss, Jena, Germany) or confocal microscope (Olympus, Tokyo, Japan), and images captured with a soft imaging systems video camera.
Results
Alterations in GABAB1R mRNA expression concomitant with ET and siRNA transfection
To investigate whether ethanol can modulate GABAB1R expression, we observed the changes in GABAB1R expression levels induced by ET on cultured GD 17.5 rat hippocampal neurons. GABAB1R mRNA levels were decreased by ET, but increased by baclofen treatment (BT) or phaclofen treatment (PT). Transfection of siRNA complementary to endogenous GABAB1R mRNA led to its efficient destruction, while β-actin levels were unchanged (Fig. 1).
Alterations in protein expression of GABAB1R, PKA-α, CaMKII and p-CREB concomitant with ET and siRNA transfection
We wished to ascertain whether ET-mediated transcriptional alterations affected protein levels in cultured GD 17.5 rat hippocampal neurons. Western blotting results showed that GABAB1R protein levels were increased by ET in vitro, but this effect was reversed by PT. BT potentiated the effect of ethanol on GABAB1R expression (Fig. 2). In contrast, no remarkable change in PKA-α level was observed under ET or PT in vitro (Fig. 3). Because PKA-α levels were unchanged upon GABAB1R knockdown, as well as BT and PT, we speculated that CaMKII might instead regulate p-CREB. Ethanol decreased CaMKII expression in GD 17.5 rat hippocampal neurons, whereas BT, PT, and baclofen plus phaclofen treatment (BPT) reversed these effects; BT, PT, and BPT significantly decreased CaMKII expression upon siRNA knockdown of GABAB1R (Fig. 4). p-CREB protein levels were decreased by ET, an effect reversed by BT, PT did not alter protein expression. We additionally probed whether ethanol-induced changes in GABAB1R expression affected its intracellular signal transduction, knocking down its expression by RNAi to that end. GABAB1R knockdown reduced all of p-CREB expression level (Fig. 5).
Co-localization of GABAB1R, p-CREB, and changes in distribution with siRNA transfection
To confirm the localization of GABAB1R and p-CREB, and to determine changes mediated by siRNA knockdown of GABAB1R, we performed immunofluorescence assays on GD 17.5 rat hippocampal neurons. GABAB1R co-localized with p-CREB in hippocampal neuronal cytoplasm and p-CREB distribution was reduced by GABAB1R siRNA (Fig. 6).
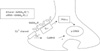
Together, these results demonstrate that ethanol can modulate phosphorylation of CREB via PKA-α or CaMKII pathway intracellular Ca2+ dependently, which is linked to GABAB1R activation in prenatal hippocampal neurons (Fig. 7).
Discussion
In this study, we have endeavored to characterize alterations to GABABR, PKA-α, and p-CREB expression in response to ET in cultured hippocampal neurons. We have additionally tested the modulatory effects of a GABAB1R agonist and/or antagonist on ET-mediated GABA-ergic signaling, as well as that of GABAB1R gene knockdown. We find that ethanol regulates GABABR expression disparately at mRNA and protein levels, suggesting that these effects are not completely attributable to changes in gene expression as previously mentioned [21].
Ethanol increased GABAB1R protein expression, but decreased that of PKA-α and p-CREB, in our study. Previous research has demonstrated that ET reduces hippocampal p-CREB protein levels in a manner reversible upon its withdrawal [22]; however, there is some contention over these findings, as other work has found that chronic ethanol treatment (CET) may decrease long-term potentiation in the hippocampus through reduction in presynaptic GABAB autoreceptor, and CET decreases GABABR expression consistent with increased GABA release [23]. Additionally, PKA and p-CREB protein expression were significantly lower in the hippocampi of model rats experiencing chronic, unpredicted, mild stress than in normal control animals [24], and alcohol-preferring rats that exhibit increased ethanol consumption likewise increase p-CREB expression selectively in the central amygdala after acute ET. Thus, it has been assumed that ethanol-induced reduction in GABAB1R levels stems from tolerance to ET in vivo [25].
We determined that p-CREB levels were decreased by ET in vitro, and that this decrease could be counteracted by BT. These findings agreed with previous reports that chronic binge ethanol administration results in neurodegeneration of the hippocampal dentate gyrus, reduced neurogenesis, decreases in p-CREB, decreased expression of pro-survival genes, and increased vulnerability to insults and neuronal death. Also, drug-induced CREB inactivation may lead to hippocampal neurotoxicity [26]. Several studies indicate that baclofen may reduce ethanol intake [14-17], and preclinical and clinical research has evaluated the potential of GABABR subtype agonists as pharmacotherapy for substance abuse, including that of ethanol. Baclofen contributes significantly to neuroprotection against ischemia in organotypic hippocampal slices [27]. However, the detailed molecular mechanisms for modulation by ethanol of GABABR signal transduction will require more thorough investigation.
To further confirm the modulatory effects of ET on GABABR, we knocked down GABAB1R expression by RNAi and examined the hippocampal responses to ethanol in vitro. The same effects were observed with BT, PT, and BPT after RNAi against GABAB1R. GABAB suppresses neurotransmitter release via inhibition of Ca2+ channels [28], and our results demonstrated a similar trend upon direct ethanol exposure. Activation of presynaptic GABABR at certain synapses can inhibit adenylyl cyclase, thereby decreasing cAMP levels and PKA activity to result in suppression of GABA release [29-31]. GABABR stimulation also signals to effectors through the G protein βγ subunit; βγ-mediated signaling includes the activation of G protein coupled inwardly rectifying potassium channels and the inhibition of voltage-gated calcium channels [32]. Since PKA-α and CaMKII can diffuse into the nucleus and phosphorylate CREB at the same serine residue, it is possible that GABABR and calmodulin kinase engage in crosstalk.
In conclusion, we studied alterations in GABAB1R and intracellular signaling factor expression upon ET in cultured rat hippocampal neurons. Ethanol reduced p-CREB levels in the presence of functional GABAB1R, but RNAi knockdown of GABAB1R or BT treatment reversed this effect. GABABR therefore can modulate p-CREB expression in the nucleolus through either PKA-α or CaMKII.






 XML Download
XML Download