

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Inhibitor of DNA binding (ID) proteins are members of the basic helix-loop-helix (HLH) family of transcription factors, but they lack a DNA-binding domain [1, 2]. ID proteins act as positive and/or negative regulators of gene expression through interactions with other HLH transcription factors in a context-dependent manner. ID proteins affect many cellular activities, including cell growth, differentiation, and apoptosis [3]. ID protein families share a highly conserved HLH domain and are similar in size between 13 and 20 kDa, but they display extensive sequence variation outside the HLH domain. It is well documented that the expression of each ID gene occurs in many regions of the developing nervous system in a complex and dynamic manner [4].
ID4 is a member of the ID protein family, and its expression is essentially restricted to the developing nervous system, whereas expression of ID1-3 is much more widespread during mouse embryogenesis [5, 6]. Early in neurogenesis, ID4 expression is prominent in the ventricular zone (VZ) of specific regions of the central nervous system (CNS), including the developing forebrain. Later, ID4 expression is apparent in the cortical plate of the telencephalon and the subventricular zone (SVZ) of the basal ganglia [5]. ID4 expression is also observed in the postnatal and adult brains [7, 8]. Earlier data indicated that ID4 is involved in glial cell development. ID4 expression is down-regulated in cAMP-induced astrocyte differentiation, and overexpression of ID4 was previously shown to induce apoptosis in an astrocyte-derived cell line [7]. In the developing rat optic nerve, the level of ID4 expression is strong in immature oligodendrocyte precursor cells, but then decreases with age and during culture [9]. Moreover, ID4 is present in both glioblastoma and oligodendroglioma specimens [10]. In human glioma cells, forced ID4 expression can drive malignant transformation by stimulating expression of cyclin E, resulting in a hyperproliferative profile, as well as jagged1, which activates Notch1 activation to drive astrocytes into a neural stem-like state [11].
Although previous studies have focused on the mechanism of glial cell proliferation during development and tumorigenesis, few studies have examined changes in ID4 expression in glial cells and its specific function under neuropathological conditions. In this sense, kainic acid (KA)-induced brain damage may be a suitable model for evaluating the function of ID4 during reactive gliosis, as administration of KA induces excitotoxic brain injury via oxidative stress. Furthermore, KA produces a highly specific pattern of neuronal loss in the hippocampus accompanied by intense reaction of both astrocytes and microglia [12, 13]. Accordingly, the aim of this study was to evaluate the temporal and cellular expression patterns as well as putative function of ID4 following excitotoxic hippocampal lesion formation and glial activation in the adult mouse brain.
Materials and Methods
Experimental animals and lesions
Male ICR mice (Samtako, Seoul, Korea) weighing 23-25 g each were used in this study. Animals were housed in a room under a controlled light/dark cycle (12 hour light/12 hour dark) at a temperature at 23℃. Food and water were available ad libitum. All animal-related procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Chungnam National University (2009-3-17). KA (Sigma, St. Louis, MO, USA) was prepared as a stock solution at 5 mg/ml in sterile 0.1 M phosphate-buffered saline (PBS, pH 7.4); aliquots were stored at -20℃ until required. Intracerebroventricular (ICV) injection of KA (0.1 µg/5 µl in PBS) was performed according to a previously established procedure [14]. Briefly, KA was injected at the bregma using a 50 µl Hamilton microsyringe fitted with a 26-gauge needle inserted to a depth of 2.4 mm. Control mice received an equal volume of saline. KA-injected animals (n=8 in each group) and saline-injected control animals (n=6 in each group) were allocated to three groups, which were sacrificed at predetermined times after KA administration.
Tissue preparation
One day, 3 days, and 7 days after KA or saline injection, mice were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and perfused transcardially with heparinized PBS, followed by 4% paraformaldehyde in PBS. Brains were removed, immersed in the same fixative for 4 hours, cryoprotected in 30% sucrose solution, embedded in tissue freezing medium, and then frozen rapidly in 2-methyl butane pre-cooled to its freezing point with liquid nitrogen. Frozen coronal sections (35 µm) were obtained using a Leica cryostat. Alternating sections were mounted on gelatin-coated slides or stored free floating in anti-freeze buffer. Sections mounted on gelatin-coated slides were stained with cresyl violet for routine histological examination. The stored free-floating sections were processed using immunohistochemical techniques, as detailed below.
Immunohistochemistry
Parallel free-floating sections were subjected to endogenous peroxidase blocking with 1% H2O2 in PBS, followed by treatment with blocking buffer (1% fetal bovine serum [FBS] in PBS and 0.3% Triton X-100 for 30 minutes) and incubation with ID4 (1 : 100, sc-491, Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing with PBS, tissues were exposed to biotinylated anti-rabbit IgG and streptoavidin peroxidase complex (Vector, Burlingame, CA, USA). Immunostaining was visualized with diaminobenzidine (DAB) and tissues mounted using Polymount (Polysciences, Warrington, PA, USA). Double immunofluorescence experiments were performed using Cy™3-conjugated anti-rabbit IgG (1 : 400, Amersham, Buckinghamshire, UK) for ID4 and Cy™2-conjugated anti-mouse IgG (1 : 600, Amersham) for glial fibrillar acidic protein (GFAP), a marker of astrocytes. All immunoreactions were observed under an Axiophot microscope (Carl Zeiss, Jena, Germany).
Western blot analysis
Hippocampi from the KA-treated (4 hours, 12 hours, 1 day, 3 days, and 7 days) and control mice were dissected and homogenized in lysis buffer (50 mM TrisCl, 150 mM NaCl, 0.02% sodium azide, 100 µg/ml phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin, 1% Triton X-100). After centrifugation, protein concentrations were determined in supernatants using Micro BCA protein assay kits; bovine serum albumin was used as a standard (Pierce Chemical, Rockford, IL, USA). Aliquots containing 20 µg of protein were resolved by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. For immunostaining, membranes were incubated in 5% skim milk in PBST (0.3% Triton X-100 in PBS) for 1 hour to block non-specific binding, followed by probing with ID4 and β-actin (1 : 5,000, Sigma, St. Louis, MO, USA) antibodies. Membranes were washed three times for 10 minutes each in PBST, followed by incubation for 1 hour with peroxidase-labeled secondary antibody (Vector) diluted 1 : 2,000 in PBST. After three more washes, immuno-labeled proteins were detected by chemiluminescence using a Supersignal ECL kit (Pierce Chemical) and Biomax Light-1 film (Kodak, New Haven, CT, USA). Three individual samples from the experimental hippocampi at each time point were used for Western blotting.
Primary astrocytes culture
Rat primary cerebral astrocytes were purified from neonatal rats according to standard procedure [15]. Briefly, a P1 Sprague-Dawley rat pup (Samtako, Osan, Korea) was decapitated in an ice-chilled dish and the brain removed. After removing the meninges, the cerebral cortex was dissected and dissociated in Hanks' balanced salt solution (Invitrogen, Carlsbad, CA, USA) with 55.5 mM glucose, 20.4 mM sucrose, and 4.2 mM sodium bicarbonate. After centrifugation, the cells were plated on poly-L-lysine-coated T75 flasks and maintained in modified Eagle's medium (MEM) containing 20% FBS, 100 µm non-essential amino acid solution, 2 mM L-glutamine, and antibiotics. After 7 days, the flasks were agitated on an orbital shaker for 12 hours at 200 rpm at 37℃, and the non-adherent oligodendrocytes and microglial cells were removed. The flasks were then trypzinized and expanded in Dulbecco's MEM growth media containing 10% FBS, 2 mM L-glutamine, and 1 mM sodium pyruvate. Under these conditions, the purity of the astrocytes population was 95%, as determined by immunofluorescence analysis using anti-OX-42 to detect microglial cells, anti-CNPase to detect oligodendrocyte contamination, and GFAP to identify astrocytes.
Creation of adenovirus expressing ID4
The replication-incompetent adenovirus was created using the Virapower adenovirus expression system (Invitro gen) as previously reported [16, 17]. Briefly, full-length cDNA was obtained using the primer pair 5'-ATGAAGGCGGTGAGCCCG and 5'-GCGGCACAGAATGCTGTC, which was subcloned into the pENT/CMV-GFP entry vector. Then, site-specific recombination between the entry vector and adenoviral destination vector pAd/PLDEST was carried out using LR clonase II (Invitrogen). The resulting adenoviral expression vector was transfected into 293A cells using Lipofectamine 2000 (Invitrogen). Cells were grown until the cytopathic effect reached 80% and then harvested for preparation of stock recombinant adenovirus. The adenovirus was amplified in 293A cells and purified with an Adeno-XTM virus purification kit (Clontech, Palo Alto, CA, USA).
Results
Histology of KA-induced hippocampus
Introduction of KA by ICV is a well-established excitotoxicity model that induces behavioral manifestations of seizures in mice as well as selective hippocampal cell death [18]. Hippocampal neuronal cell death was measured by cresyl violet staining. After KA administration, characteristic loss of pyramidal neurons in the CA1 and CA3 subfields of the ipsilateral hippocampus was observed from 1 day post-injection in mice (Fig. 1A2, 1A3), whereas no obvious neuronal loss was observed in the ipsilateral hippocampus of PBS-injected mice (Fig. 1A1). Pyramidal cell degeneration was more apparent in the CA3 region than the CA1 region, although cell death was also observed to a lesser extent in the CA1 region. Glial reactivity with small nuclei was increased in the hippocampus from day 1 to day 3 (Fig. 1A4-6).
ID4 expression in astrocytes following excitotoxicity
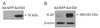
Although ID4 immunoreactivity (IR) was not observed in the control, it did occur in small glial cells in the vicinity of the hippocampus 1 day after KA treatment. Incidence of ID4 IR in cells increased significantly and was scattered throughout the CA1 region 3 days after KA treatment (Fig. 1B1-3). At higher magnification, ID4 IR evidently induced star-shaped morphology in activated glial cells in association with brain injury (Fig. 1B4-6). We performed double labeling experiments with GFAP, an astrocyte marker, to confirm which cell types were ID4 IR cells. ID4 IR cells co-localized with GFAP IR cells (Fig. 2). These results demonstrate that ID4-positive cells were predominantly activated astrocytes in the hippocampi of KA-treated mice. Western blot analysis showing the relative level of ID4 expression in KA-treated mice further confirmed the temporal pattern of ID4 IR observed in the immunohistochemical studies. Anti-ID4 antibody detected a single band of 18 kDa. The level of ID4 in the hippocampus began to increase on day 1 post-lesion and reached a maximum on day 3. Subsequently, the amount of ID4 decreased until 7 days post-lesion (Fig. 3).
Proliferative role of ID4 in astrocytes
Previous studies demonstrated that ID4 induces proliferation and differentiation in the CNS [19-22]. To confirm the effect of ID4 on proliferation, we constructed a recombinant adenovirus expressing ID4 (Ad/ID4). In Ad/ID4-transduced primary astrocytes, expression of ectopic ID4 was detected by Western blotting (Fig. 4A). Overexpression of ID4 resulted in a remarkable increase in cell number compared with the control group (Ad/GFP) (data not shown). To measure the degree of proliferation, we subjected the AD/ID4-transduced primary astrocytes to immunoblotting with a cell proliferation marker, Ki-67, which is a cell cycle-associated protein expressed in G1 phase through the end of M phase. This result confirms that ID4 mediated cell proliferation in astrocytes (Fig. 4B).
Discussion
This study examined the expression of ID4 in the excitotoxically damaged mouse hippocampus. We observed that ID4 was expressed in astrocytes following excitotoxically hippocampal neuronal death. The majority of these cells corresponded to reactive astrocytes based on their antigenic properties and morphological appearance. To our knowledge, this is the first report of ID4 induction in activated astrocytes during KA-induced excitotoxic brain injury. Previous works have shown that ID genes are highly expressed with distinct and overlapping patterns in the nervous system during development and then decreased in expression in later stages [5, 22]. In this study, we also found no ID4 IR in the normal adult mouse brain. However, expression of ID4 increased in the astrocytes after KA-treated brain injury.
It has been well reported that ID4 plays an important role in cell proliferation, differentiation, and tumorigenesis. ID4 is a transcriptional target of p53, and depletion of mutant p53 protein severely impairs ID4 expression in proliferating tumor cells. The protein complex mutant p53-E2F1 assembles on specific regions of the ID4 promoter whereupon it positively controls ID4 expression. ID4 protein binds, stabilizes and enhances the translation of mRNAs encoding the pro-angiogenic cytokines, such as interleukin 8 and growth-regulated oncogene (GRO)-alpha. Therefore, ID4 increases the angiogenic potential of cancer cells [23]. In addition, ID4 is highly expressed in the glioblastoma multiforme, which is the most common and aggressive type of primary astrocytoma, and plays an integral role in the transformation of astrocytes [11]. Tumor-derived cell cultures expressing elevated levels of ID4 produce enlarged xenografts in immunosuppressed mice that vascularize better than corresponding control tumors. This suggests a novel pro-angiogenic function for ID4 in the growth of glioblastoma xenografts [24]. In this sense, we tried to identify a putative role for ID4 in astrocytes as gain of function using an adenoviral vector. Overexpression of ID4 induced the proliferation of astrocytes. Based on these results, we speculate that ID4 plays a role in the proliferation of astrocytes following excitotoxic hippocampal neuronal death.
Previously, glial activation was observed in scattered glial cells in whole hippocampus [12, 13]. However, we observed that ID4 was expressed in astrocytes, especially the CA1 layer, following excitotoxic hippocampal neuronal death. Except for olfactory bulb neurogenesis, little is known about the role of neural precursor cells residing in the SVZ of ventricle walls [25]. Under certain pathological conditions, a small number of these cells have been shown to contribute to regenerative neurogenesis outside the classical neurogenic regions of the brain [26], namely, the olfactory bulb and hippocampal dentate gyrus. In another study, when pyramidal neurons in the hippocampal CA1 area were eliminated by focal ischemia, restorative neurogenesis was achieved by high-dose administration of fibroblast growth factor 2 and epidermal growth factor [27]. This regeneration originated from cells migrating into the CA1 region from the ventricle wall, in particular from the caudal SVZ. In addition, prolonged status epilepticus has been reported to recruit SVZ glial progenitors to the CA1 and CA3 regions [28].
ICV KA administration results in the degeneration of a vast majority of CA3 pyramidal neurons and a significant fraction of dentate hilar neurons. However, both CA1 pyramidal neurons and dentate granular cells are unaffected by ICV KA administration. Degeneration of CA3 pyramidal neurons leads to significant deafferentiation of the dendrites of CA1 pyramidal neurons. Thus, diffuse and pervasive synaptic reorganization and hyperexcitability occur in the CA1 subfield after ICV KA-induced CA3 lesion formation [29]. It is important to stress that these changes in astrocytes have been observed in the absence of overt neuronal degeneration. Therefore, our finding that ID4 was expressed following excitotoxic hippocampal neuronal death in astrocytes of the CA1 layer, which were previously SVZ glial progenitors that migrated to the CA1 from the caudal SVZ, implies that proliferation occurred. In summary, we demonstrated that KA-induced excitotoxic neuronal death and glial activation induces ID4 expression in astrocytes, and ID4 plays an important role in astrocytic proliferation in response to excitotoxin in vivo.



 XML Download
XML Download