

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Mitochondrial genomes (mtDNA) and the complex genetics of mtDNA disorder
Mitochondria are ubiquitous subcellular organelles, present in all eukaryotic cells. Although they are involved in several central metabolic pathways that are crucial for cellular function, their primary function is the conversion of food energy into chemical energy (ATP) through the oxidative phosphorylation (OXPHOS), which can be used to drive the important cellular reactions (Green & Reed, 1998; Saraste 1999). Mitochondria are composed of two membranes with which two discrete compartments, the internal mitochondrial matrix space and the narrow intermembrane space, are structured (Frey & Mannella, 2000; Perkins et al., 2009). The outer membrane is permeable to molecules smaller than 5 kDa, making the intermembrane space chemically equivalent to the cytosol with respect to small molecules (Weissig et al., 2004). The inner membrane contains a high content of membrane proteins including components of the electron transport chain complexes as well as ATP synthase and a whole variety of transport proteins. Due to a unique lipid composition, the inner mitochondrial membrane is highly impermeable to any cellular molecules and this impermeability can generate an imbalance in the distribution of protons (H+) between the mitochondrial matrix and the intermembrane space. The chemical imbalance, i.e. proton gradient, is used for the driving force of ATP synthesis in mitochondria.
Mitochondria are generally thought to have arisen from an intracellular bacterial symbiont of the first ancestral eukaryotic cells, which presumably provided most of the energy metabolism for this symbiotic pairing (Gray et al., 1999). During the evolution of symbiotic relationship between these proto-bacteria and early eukaryotic cells, most of the genetic information from the circular proto-bacterial genome (proto-mitochondrial genome) was either lost or was transferred to the nuclear genome of the host eukaryotic cells. Because the evolution of mitochondrial and nuclear genomes in various eukaryotic cells has been thought to occur simultaneously, the mitochondrial genomes of different eukaryotic lineages differ in size, gene content and even the genetic code that they use (Gray et al., 1999). The modern mammalian mitochondrial genome is a circular DNA molecule that has been reduced to ~16.5 kb in size and encodes genes for only thirteen protein products, all of which are critical components of the electron transport chain, as well as two ribosomal RNAs and 22 transfer RNAs that are required for the mitochondrial translation system. All of the other genes needed for the biogenesis, maintenance and regulation of this organelle are encoded in the nucleus. Each of the genes encoded in the mitochondrial genome, however, remains critical for normal mitochondrial function (Wallace 1999).
The human mitochondrial genome is a circular double stranded DNA molecule with a size of 16,569 bp (Fig. 1). The mtDNA has no intron but retains compactly arranged 37 genes (13 proteins, 22 tRNAs and 2 rRNAs) critical for producing energy through OXPHOS. Major noncoding regions in the mtDNA genome involve the D-loop sequence and the origin of L-strand replication (OL), which controls mtDNA transcription and replication within mitochondria. The 13 protein-coding genes encode subunits of the OXPHOS enzyme complexes. Seven polypeptides (ND1~ND6 and ND4L) are involved in subunits of complex I, one (cytochrome b) is part of complex III, three (COXI~COXIII) are in subunits of complex IV and two are part of ATP synthase (ATP6 and ATP8) (Scheffler 2001). The remaining 24 genes (22 tRNAs and 2 rRNAs) encode the translational machinery of the mitochondrial genome itself. The mitochondrial genetic codes (mt) are similar to the nuclear codes (nu) but not exactly identical with them. Four codons are different in mammalian nuclear and mitochondrial translational system: AUA=Met (mt), Ile (nu); AGA, AGG =Stop (mt), Arg (nu); UGA=Trp (mt), Stop (nu). In addition to the different codon usages, mitochondria have some unique characteristics which reflect that the organelles are evolved from endosymbiotic proto-bacteria. The mitochondrial gene expression system still carries hallmarks of its bacterial ancestor (Wallace 1999; 2007). For example, an N-formylmethionyl-tRNA (fMet-tRNA) which is involved in the initiation of protein synthesis in bacteria is employed as initiator of protein synthesis in mitochondria (Galper & Darnell, 1969; Epler et al., 1970).
It is now well known that mutations in the mitochondrial genome can cause a wide range of human diseases (Wallace 1999). Mutations in human mitochondrial genome were first reported in 1988. Wallace et al. (1988) demonstrated that a nucleotide change in a mitochondrial DNA energy production gene (NADH dehydrogenase subunit 4, ND4) can result in a neurological disease, Leber's hereditary optic neuropathy (LHON). Furthermore Holt et al. (1988) found deletions in muscle mitochondrial DNA from patients with mitochondrial myopathies. Within a decade, over 100 pathogenic point mutations as well as multiple deletions and rearrangements involving essentially every mitochondrial gene had been described (DiMauro & Schon, 2001). Human diseases related with defective mitochondrial function exhibit a broad array of clinical problems and often involve tissues that have high energy requirements, such as heart and other muscle and the endocrine and nervous systems (Wallace 1999). Typically a single mitochondrial mutation leads to a complex medical syndrome, with clinical features that may include such problems as muscle deprivation and frailty, vision loss, ataxia, stroke-like episodes, cardiomyopathy, seizures, and hearing loss. The close causal relationship between accumulation of mtDNA mutations and age-related functional loss is evident in a mouse model of hyper-mutated mitochondrial DNA that was induced by a proof-reading-deficient version of the mitochondrial DNA polymerase γ (Trifunovic et al., 2004; Edgar et al., 2009). These mutants exhibit accelerated accumulation of mtDNA mutations, increase of heart weight, enlarged mitochondria and the phenotype of kyphosis, alopecia, osteopenia, loss of body mass, anemia, lipodystrophy, cardiomyopathy and reduced fertility. Heart muscle tissues from these mtDNA-mutator mice have a mosaic pattern, with cytochrome c oxidase deficiency, which is normally found in the ageing human heart (Trifunovic et al., 2004). In addition to those diseases in which a mitochondrial genome mutation is the primary cause of the disease, there is also a growing body of evidence that abnormal mitochondrial function plays a role in the pathology of many other diseases as well, including Huntington's Disease (Panov et al., 2002), Alzheimer's Disease (Castellani et al., 2002) and even cancer (Clark et al., 2002).
Although the mammalian mtDNA genome has been amenable to many of the standard molecular techniques used to study the nuclear genome (Yoon & Koob, 2003), no practical method has yet been developed to introduce mutagenized mtDNA directly and stably into the mitochondria of a mammalian cell. Genetic manipulation of mitochondrial genome and its gene products is also one of the approaches under investigation for the treatment of mtDNA disorders. Yeast mitochondria can be transformed with exogenous DNA using a biolistic procedure (Fox et al., 1988; Johnston et al., 1988), but to date this method has been limited to the transformation of mitochondria in yeast and closely related organisms. This technical roadblock is preventing many important experiments from being performed in the field of mammalian mitochondrial genetics, and as a result mitochondrial transformation has long been viewed as the "holy grail" of this field (Shoubridge 2000).
In this review we conduct a breif overview of the current progress in manipulating mammalian mitochondrial genomes using mtDNA delivery systems and other approaches. The ability to modify the mitochondrial genomes would provide a powerful tool to create mutants with which many crucial experiments can be performed to advance our knowledge in the field of mitochondrial genetics as well as to develop ways of treating human mtDNA diseases. At the end of this review, we summarize our own efforts for introducing engineered mtDNA constructs into the mitochondria of living cells through bacterial conjugation.
Current approaches of modifying mitochondrial genomes in mammalian cells
Mammalian mitochondrial transformation with exogenous DNA molecules is a complex field of mitochondrial research and is still in early stages. Although many efforts were undertaken during the last decade and promising in vitro experimental results were reported, no one can generate transformed cells with modified or engineered mitochondrial genomes in their mitochondrial networks. Introducing exogenous DNA including re-engineered mitochondrial genomes has proven to be more challenging than strategies directed at the nuclear genome. In the following sections, we discuss different approaches towards the modification of mitochondrial genomes through direct subcellular disposition of nucleic acids and/or modifying proteins.
Transferring nucleic acids directly by conjugating with mitochondriotropic molecules
Lipophilic cationic compounds have amphiphilic characteristics with delocalized positive charges. Positively charged hydrophobic molecules have tendency to form amphipathic helices which could pass through mitochondrial membranes and acculmulate in the mitochondria matrix (Hashimoto et al., 1984; Rottenberg 1984). Because mitochondria have highly negative membrane potential, cationic molecules such as rhodamin 123 (Chen et al., 1982) and ethidium bromide (Nass 1970) which are commonly used to visualize mitochondria and to deplete mtDNA, respectively, are attracted to negatively charged mitochondria.
One of lipophilic monocation, triphenylphosphonium (TPP), can pass through phospholipid bilayers and accumulate in negatively-charged mitochondrial matrix. This unique characteristic of TPP is specifically used to deliver small therapeutic molecules such as antioxidants ubiquinone and vitamin E to mitochondria (Smith et al., 2003; Ross et al., 2006). TPP-facilitated delivery of peptide nucleic acids (PNA) has also been investigated in an antisense mitochondrial gene therapy system (Muratovska et al., 2001). PNA is a synthetic DNA-like polynucleotide in which each nucleotide is connected by a polyamide backbone (peptide bond), rather than by deoxyribose phosphodiester subunits (Nielsen et al., 1991) and can stably form double strands with complementary DNA or RNA molecules. PNA conjugated with TPP can cross the mitochondrial membranes and selectively suppress mtDNA replication by binding to the complementary DNA sequences. An in vitro model has shown that TPP conjugated PNA exclusively hampers the replication of mtDNA containing the A8344G point mutation, which causes the human mtDNA disease 'myoclonic epilepsy and ragged red fibrs' (MERRF) but not the wild-type sequence that differs at a single nucleotide position (Muratovska et al., 2001). Since mitochondrial genomes are maintained as multicopy status and wild type and mutant mtDNA coexist in many patients with defective mitochondrial function, selective inhibition of mutant mtDNA replication would be required for the treatment of the mitochondrial disease. Therefore therapeutic potential of this antisence PNA gene therapy must be the sequence-specific and selective inhibition of mutant mtDNA in mitochondria. However, in an in vivo model with the MERRF mtDNA, TPP-PNA conjugates did not selectively inhibit replication of mutant mtDNA even though they were accumulated several hundred-fold into the mitochondria. One possible explanation is that any single-stranded mtDNA present during mtDNA replication is unavailable for binding to complementary PNAs. This could occur because binding is prevented by proteins associated with the DNA, or because the lifetime of the single-stranded DNA is insufficient (Muratovska et al., 2001). This hurdle may be overcome by designing PNA oligomers that can bind specific double-stranded DNA sequences to form a stable triple helix.
DQAsomes are mitochondriotropic bolasomes that are derived from the compound dequalinium (Weissig et al., 1998). Dequalinium is a dicationic compound resembling bolaform electrolytes and is a symmetrical molecule with two charge centers separated at a relatively large distance. This symmetric bipolar compound can self-associate into mechanically very stable spherical monolayer aggregates (called DQAsomes) upon sonication in aqueous medium. DQAsomes have been observed to migrate and localize with mitochondria within living cells (Weissig et al., 1998; Weissig & Torchilin, 2000; D'Souza et al., 2003). These liposomal-like vesicles are capable of binding plasmid DNA and form DQAsome-DNA complexes in which the plasmid DNA is protected from nucleases. DQAsomes have been employed to deliver both oligonucleotides and plasmid DNA conjugated with a mitochondrial targeting sequence (MTS) to mitochondria of living cells (D'Souza et al., 2005). Although DQAsome-DNA complexes can approach to mitochondria inside the living cells and colocalize with a mitochondrial-specific dye, MTS conjugated with DNA was not sufficient to assist DNA internalization into the matrix space (D'Souza et al., 2005). Nevertheless DQAsomes may be used to deliver functional recombinant mtDNA construct to mitochondria. As the full recombinant mtDNA clones are now available and conventional drug resistance proteins (aminoglycoside phosphotransferase (NeoR) and hygromycin B phosphotransferase (HygR)) present in mitochondria are functional and confers resistance to high levels of G418 (aminoglycoside antibiotic) and hygromycin (Yoon & Koob, 2003; 2008; Yoon et al., 2009), DQAsomes-mtDNA complexes with and without a drug resistance gene could serve as effective mediators for mtDNA transfer into the mitochondrial network of wild type tissue culture cells and mtDNA-less ρ0 cells, respectively. DQAsomes deliver recombinant mtDNA construct at the site of mitochondria in cells, and by an effective DNA import mechanism such as the mitochondria natural import competence (Koulintchenko et al., 2006), the mtDNA construct may be introduced into mitochondrial matrix where the DNA can be properly replicated and transcribed. Weissig group also investigated mitochondriotropic liposomes by linking the TPP cation into conventional liposomes (Boddapati et al., 2005). The phospholipid vesicles (liposomes) are rendered mitochondrial-specific by modifying the liposomal surface with TPP and the modified liposomes seem to accumulate at or near the site of the mitochondria. However, these mitochondriotropic liposomes show high level of cytotoxicity when applied in tissue culture cells (Boddapati et al., 2005). The cytotoxicity may have resulted from direct contact or association of the mitochondriotropic liposomes with mitochondrial membranes and consequently this interaction may induce channel formation, which initiates an apoptotic chain reaction from the damaged mitochondria.
Transferring nucleic acids through conjugating mitochondrial targeting sequences (MTS)
Among ~1,500 mitochondrial proteins which participate mitochondrial biogenesis including the oxidative phosphorylation, only 13 proteins are encoded from the mitochondrial genome and the rest of the mitochondrial proteins are expressed from the nuclear genome and actively transported to the mitochondria. These mitochondrially transported proteins contain leader sequences which direct molecules to the mitochondria. Typically matrix targeting sequences are 10~80 amino acids in length, are generally have the ability to form amphipathic helices (von Heijne 1986; Mukhopadhyay et al., 2003). The MTS is recognized by the mitochondrial import complexes (translocases of the outer membrane (TOM) and the inner membrane (TIM)) and mediates mitochondrial localization, and subsequently deliver mitochondrial proteins to the matrix compartment. Thus, the MTS and mitochondrial protein import complexes have been explored for nucleic acids import to mitochondria.
In an early study, mitochondrial import of a single-stranded or double-stranded 24-bp piece of DNA was shown by covalently conjugating the DNA with the MTS of yeast cytochrome oxidase (Vestweber & Schatz, 1989). The Mitochondrial import of chemically synthesized signal peptides and their conjugates to small single- and double-stranded DNA have also been investigated using isolated rat liver mitochondria (Seibel et al., 1995; Lu & Beavis, 1997). Among these experiments, Seibel et al. (1995) confirmed that the leader sequence conjugated to double stranded DNA molecule of a 17 or 322 bp in size was imported to the mitochondrial matrix by utilizing the protein import pathway. They also suggests that the translocation of MTS-conjugated DNA is independent from the size of its passenger DNA and that comparatively large DNA molecules over 322 bp could possibly be imported to mitochondria by attaching to MTS. The mitochondrial targeting sequence has also been conjugated to a PNA complementary to a portion of the mtDNA sequence (Flierl et al., 2003). This targeting peptide-PNA (MTS-PNA) conjugate was annealed to an oligonucleotide complementary to the PNA sequence and then the complex was transferred into the cytosol using commercially available cationic liposome formulations. This procedure efficiently delivered the oligonucleotides into the mitochondrial matrix. In another approach, an oligonucleotide was coupled covalently to a mitochondrial targeting peptide sequence and then the MTS-oligonucleotides were encapsulated with a cationic liposome (Geromel et al., 2001). These modified MTS-oligonucleotides complexed to liposomes have been shown to enter the cytoplasm of cells, dissociate from the complexes, and then penetrate into the mitochondria of primary cultured cells.
The use of MTS is considered to be the most promising candidate for the delivery of DNA to mitochondria when complexed with cationic liposomes including DQAsomes (D'Souza et al., 2005). However, the experiments performed previously used oligonucleotides or relatively small linear DNA fragments to transport into mitochondria. Although MTS peptides actively contribute to a mitochondrial targeting effect and subsequently mediate the transport of the complex across the mitochondrial membranes, the remaining question is that whether conjugated MTS peptide could deliver a large circular DNA molecule such as recombinant mtDNA constructs to mitochondria. This question should be answered because a mitochondria-specific delivery system requires an efficient delivery of circular DNA to mitochondria for the purpose of modifying or manipulating DNA in mitochondria of living cells.
Transferring nucleic acids using polyethylenimine (PEI) polymer
Direct DNA delivery to the mitochondrial matrix has been investigated with MTS-conjugated PEI (MTS-PEI) (Lee et al., 2007). PEI is a polymeric transfection agent that has been widely used for gene delivery to the nucleus including circular plasmid vectors. It has a high positive charge density and therefore PEI and DNA can create condensed positively charged particles which, in turn, bind to anionic cell surface residues and are brought into the cell via endocytosis. MTS-PEI has been shown to form complexes with DNA equally well as shown in PEI itself. When applied in a living cell, MTS-PEI can transport DNA into mitochondrial sites, indicating that the DNA delivery to mitochondria using MTS-PEI could be a feasible approach. However several important questions regarding this PEI polymer as a mitochondrial delivery vector remain to be answered. First, DNA should be released from the MTS-PEI complexes when they arrive to the mitochondrial site. The dissociation of the MTS-PEI/DNA complexes allows DNA to be freed and allows it to be imported into the mitochondria. In spite of many efforts to address this question, it is not yet clearly demonstrated which step enables the release of DNA from the PEI/DNA complexes. Second, it has not been addressed whether MTS-PEI/DNA complexes are internalized into the mitochondrial matrix or bound to the surface of the mitochondrial membrane. If the MTS-PEI binds to mitochondrial membrane and induces channel formation on the outer mitochondrial membrane, cytochrome c may be released into the cytosol, which would force cells to enter apoptosis. If this is the case, subtle control of working concentration of PEI complexes should be determined to minimize cytotoxicity to cells.
Transferring recombinant mtDNA constructs through electroporation and natural import competence
Several pioneering efforts of introducing exogenous circular plasmid DNA physically into mitochondria have been demonstrated. Collombet et al. (1997) first described the transfer of small 7.2-kb recombinant plasmids into isolated mouse mitochondria by electroporation and proved the functional integrity of the electroporated mitochondria using enzymatic assays. Subsequently Yoon & Koob (2003) optimized the electroporation conditions for introducing a full mitochondrial genome construct using mitochondria isolated from mtDNA-deficient mouse ρ0 cells and demonstrated the biological activity of the recombinant mouse mitochondrial genomes by detecting in organelle RNA expression through an RT-PCR assay specific for transcripts from the transferred DNA. The basic idea of these approaches is that the modified mtDNA construct is introduced into isolated mitochondria by electroporation and then the electroporated mitochondria are transferred into zygotes or tissue culture cells through a microinjection technique to transfer engineered mtDNA constructs. Current microinjection technique allows for the introduction of isolated mitochondria into a living cell. The injected mitochondria can lead to a rapid replacement of the endogenous mitochondrial DNA in cells (King & Attardi, 1988; 1989). However, the harsh experimental conditions required for electroporation appear to cause irreversible damage to the structural integrity of the mitochondria and kill mammalian cells when applied to whole cells (Yoon & Koob, 2005). As an interesting alternative method, the natural import competence of isolated mammalian mitochondria for DNA has been demonstrated (Koulintchenko et al., 2006). This study showed that naked DNA can be spontaneously imported into the isolated mitochondria when the DNA and the mitochondria are mixed and that the imported DNA could be used as a substrate for DNA synthesis and promoter-driven transcription, with the resultant polycistronic RNA being processed and excised mt-tRNA matured (Koulintchenko et al., 2006). Although in vivo phenomenon of this natural competence of naked DNA into mitochondria has not been reported, the authors noted that in vivo DNA transport to mitochondria may be feasible in combination with cationic liposomes such as DQAsomes (see above). Cationic liposomes associate with DNAs and condense them into DNA-liposome complexes, which are able to access to cytosol and localize in mitochondria. If the DNA is released efficiently from these DNA-liposome complexes near the mitochondrial site, the freed DNA might spontaneously be imported into the mitochondrial networks through this natural import competence (Koulintchenko et al., 2006).
Modulating mtDNA content by mitochondrially targeted restriction endonucleases
Patients with mitochondrial DNA disease usually harbor a mixture of mutant and wild-type mtDNA, which is termed heteroplasmy. The clinical features of the mtDNA disease depend on the percentage of mutant mtDNA (the 'mutation load') in vulnerable tissues. Several studies have shown that there is a threshold whereby a certain level of mutant mtDNA is necessary before the disease becomes biochemically and clinically apparent (Thorburn & Dahl, 2001; DiMauro & Schon, 2003). To modulate mutant mtDNA levels in living cells, mitochondrially targeted restriction endonucleases have been used (Srivastava & Moraes, 2001; Tanaka et al., 2002). Based on the presence of mtDNA in a heteroplasmic state, Srivastava & Moraes (2001) and Tanaka et al. (2002) demonstrated experiments manipulating mutant mtDNA content by a mitochondrially targeted PstI and SmaI (MTS-PstI and MTS-SmaI), respectively. Mutant mtDNA is selectively destroyed or its replication is inhibited by the endonuclease and thereby only wild-type mtDNA propagate in the mitochondria. Recently, a heteroplasmic T8993G mutation which is a cause for the NARP and MILS syndromes, has also been selectively removed by the mitochondrially targeted R.XmaI restriction endonuclease (Alexeyev et al., 2008). This restriction endonuclease approach, however, is relatively limited in terms of the range of mutations that can be targeted. Not all mutations will produce a restriction site and even though a site is generated, it may not be unique, and hence would digest wild-type mtDNA molecules. To solve this problem, the selective degradation of mutated mtDNA molecules has been tested using zinc-finger proteins (ZFPs) (Minczuk et al., 2008). Zinc finger technology allows for the engineering of zinc-finger proteins that can bind any predetermined DNA sequence (Kim et al., 1996; Wu et al., 2007). Fusing a particular ZFP to a nuclease domain creates a zinc-finger nuclease (ZFN) that can cleave DNA adjacent to the specific ZFP-binding site. By designing a single-chain quasi-dimeric ZFN with predetermined DNA binding domain, ZFN could recognize a pathogenic point mutation in mtDNA, selectively cleave and eliminate the mutated mtDNA and thereby increase the proportion of wild type mtDNA in the cells (Minczuk et al., 2008). ZFN technology therefore could provide a powerful tool to modulate heteroplasmy for treating disease caused by mtDNA point mutations.
Mitochondrial gene replacement therapy using protein transduction
Recently, a novel protein transduction technology (protofection) that would allow insertion and expression of mitochondrial genomes into living cells has been reported (Khan & Bennett, 2004; Keeney et al., 2009). Protofection uses recombinant human mitochondrial transcription factor A (TFAM) engineered with an N-terminal protein transduction domain (PTD) followed by the MTS to deliver mtDNA cargo to the mitochondria of living cells. Since TFAM is the major mtDNA-binding protein with two high-mobility group (HMG) domains, it binds to and organizes mtDNA into a mitochondrial nucleoid structure, which is necessary for mtDNA transcription and maintenance (Parisi & Clayton, 1991; Alam et al. 2003). Therefore the MTD-TFAM (MTD = PTD+MTS = mitochondrial transduction domain) recombinant protein would bind mtDNA by interacting with TFAM and rapidly transport it across plasma membranes to mitochondria with the assistance of MTD. For therapeutic purposes, protofection technology has been tested in Parkinson's disease (PD) cybrid model cells by combining MTD-TFAM with wild type mtDNA (Keeney et al., 2009). Typically PD cybrids show impaired respiration and a reduced mtDNA level, but when the PD cybrid cells were treated with the MTD-TFAM/wild type mtDNA mixture the cells showed increased mtDNA copy numbers, mtDNA-derived RNA levels, TFAM and electron transport complex proteins, cell respiration, and physical mitochondrial movement, which suggested the possibility of therapeutic application of protofection technology to mtDNA diseases. However this approach requires additional experiments to identify the molecular mechanisms whether the therapeutic effect is derived from the wild type mtDNA that was exogenously transferred and this protein transduction technology should be reproducible by other investigators.
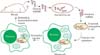
A novel approach to re-engineer mitochondrial genomes through bacterial conjugative transfer of plasmid DNA into mitochondria
As for a unique approach, we have transferred exogenous plasmid DNA successfully into the mitochondrial matrix by means of conjugation between bacteria and mitochondria (Yoon & Koob, 2005). Because conjugation is a relatively gentle process, this type of procedure would enable the transfer of large DNA constructs into mammalian mitochondria without damaging their structural integrity. Typically the bacterial conjugation process is driven entirely by molecular machinery in the donor bacterial cell (Waters 1999) and it is now well known that a broad range of cell types can serve as the DNA recipient, including yeast (Heinemann & Sprague, 1989; Nishikawa & Yoshida, 1998), plants (Piers et al., 1996) and even mammalian cells (Kunik et al., 2001; Waters 2001). Because mitochondria arose from an intracellular bacterial symbiont of the first ancestral eukaryotic cells (Gray et al., 1999) and have inner and outer membranes that are generally similar to many bacteria, it is perhaps not surprising that the bacterial conjugation system can transfer DNA constructs into mitochondria as well (Yoon & Koob, 2005).
In order to determine whether bacterial conjugation could serve as an effective means of transforming mitochondria, an assay system was designed to specifically detect DNA that was transferred into the matrix of structurally intact mitochondria (Yoon & Koob, 2005). In this assay, the transferred DNA is transcribed by an RNA polymerase that is present only in the mitochondrial matrix and these transcripts are specifically detected by RT-PCR. Any DNA or RNA outside of the mitochondria is eliminated by specifically lysing the E. coli donor cells and adding high levels of DNase and RNase prior to in organelle RNA isolation. Although this assay could perhaps have been performed by placing a mitochondrial promoter on the mobilizable DNA construct and relying on the endogenous mitochondrial RNA polymerase to generate transcripts from transferred DNA, we instead chose to use the phage T7 transcription system because T7 RNA polymerase can provide robust RNA transcription from the DNA template and is well-characterized through many experiments. In order to adapt this conjugative DNA transfer system to the transformation of mitochondrial networks within living cells, we employed a conjugative E. coli strain that is capable of invading the cytoplasm of mammalian cells. The overall experimental scheme is shown in Fig. 2. Lim et al. (2008) have shown that the bacterial conjugation machinery is functional in the cytoplasm of mammalian cells. Specially constructed invasive E. coli was able to escape from the phagocytic vacuole and transfer a conjugative plasmid at high frequency to a recipient E. coli that was invaded separately (Lim et al., 2008). Therefore a conjugative and invasive strain of E. coli could be used as a delivery vehicle for transferring DNA into the mitochondrial networks of mammalian tissue culture cells.
Making a non-replicating but metabolically active (conjugative) E. coli strain
Since this experimental system requires invasive nature of E. coli, we employed a clinical isolate of an enteroinvasive E. coli (EIEC). This strain can invade and replicate in the cytoplasm of both mouse and human tissue culture cells when we tested the EIEC strain by gentamicine protection assay (Sinai & Bavoil, 1993) (Fig. 3). Typically, gentamicin applied to the culture medium kills any bacteria outside of the cultured cells and thus only the E. coli invading the cytoplasm are protected from the antibiotic. The tissue cultured cells are then lysed and the lysates are spreaded on LB medium to verify the viable E. coli (Fig. 3). To enable this strain to serve as a conjugative DNA donor, we introduced a conjugative "helper plasmid" into this strain and confirmed that the resulting strain could transfer DNA into other E. coli strains. This initial strain, however, quickly replicates in the cytoplasm of tissue culture cells and kills them (see Fig. 3, upper panel). To alleviate this problem, we removed the gene of the E. coli chromosomal replication initiation protein dnaA (Sutton & Kaguni, 1997) and transferred it onto a plasmid that replicates at a low temperatures (30℃) but not at a high temperature (42℃) (Fig. 4A). This system allowed us to culture this strain at a low temperature and then to shift to a high temperature in order to generate a population of non-replicating but metabolically active (conjugative) daughter cells. These non-replicating and conjugative daughter cells could be readily separated from any remaining replication-competent parent cells. The replicating E. coli cells could be selectively removed from the culture by lysing the cells using antibiotics such as ampicillin. Beta-lactam antibiotics including ampicillin exert their bactericidal effects by inhibiting the cross-linking step (transpeptidation) of bacterial cell wall biosynthesis, resulting in cell lysis (Waxman & Strominger, 1983). During this incubation, therefore, only the non-replicating E. coli remains intact and the replicating one is lysed by ampicillin. When we tested the replication ability of ampicillin-treated (+Amp) or non-treated (-Amp) E. coli on LB plates (Fig. 4B, plates 1 and 2), no colony formed in the ampicillin-treated cultures (Fig. 4B, plate 2). When we tested the conjugation ability of the ampicillin-treated EIEC by mating them with recipient E. coli DH5α, we found that the recipient cells were able to grow on selection medium (Fig. 4B, plate 4), indicating the non-replicating EIEC is capable of donating plasmid DNA containing a selectable marker to a recipient E. coli strain. These results revealed that a population of non-replicating but metabolically active (conjugative) E. coli can be obtained using temperature and antibiotic treatment.
Invasion of non-replicating E. coli into mammalian tissue culture cells
Since a population of non-replicating E. coli cells can be separated by controlling temperature and treating ampicillin, we next tested whether these non-replicating forms of E. coli could still actively invade the cytoplasm of tissue culture cells (Fig. 4C). Because these E. coli could not form any colonies, we assayed the invasion of non-replicating E. coli by labeling them with GFP and visually confirmed that they were present in the cytoplasm of the cells in the invaded cultures. Note that the non-replicating E. coli are more elongated than the replicating forms due to continued cell growth in the absence of genomic DNA replication. The size of the E. coli shown in Fig. 4C, which is roughly four times the length of the rapidly dividing cells, is typical of the cell length in non-dividing cultures. However the number of non-replicating E. coli invading the cytoplasm decreased compared to the wild type EIEC strain. We, however, significantly improved the number of E. coli that can invade the cytoplasm of tissue culture cells by adding the invasin gene from Yersinia pseudotuberculosis (Grillot-Courvalin et al., 1998) into our conditionally replicating EIEC bacterial strain (Fig. 4D). The invasin surface protein binds to cell-adhesion proteins on the tissue culture cells and enhances phagocytosis. Fig. 4D shows a single tissue culture cell photographed after invasion with a GFP labeled, non-replicating population of these invasive E. coli. Nearly 100% of invasion efficiencies was achieved with this modified strain, while still retaining viability in most of the invaded tissue culture cells using ampicillin to eliminate all of the replicating EIEC bacteria from the cultures prior to invasion.
In the bacterial conjugative DNA transfer approach shown in Fig. 2, we used E. coli as an mtDNA delivery vector after providing invasive and conjugative characteristics. Since the invasiveness of this E. coli strain has been evaluated, the conjugation process of the strain between mitochondria and E. coli should be defined in a cytoplasmic environment of cells. It would be informative to use T7 tissue culture cell lines in which T7 RNA polymerase is targeted to the mitochondria and a mobilizable mtDNA construct that contains a T7 promoter along with a fluorescent marker (GFP or DsRed) gene. Once the invasive, non-replicating and conjugative EIEC strains have delivered the T7 constructs into the mitochondria of T7 cell lines in which the T7 RNA polymerase is already targeted, we may be able to observe the fluorescence expression from these mitochondrial networks through the strong T7 expression system. Because single stranded DNA is typically transferred by bacterial conjugation, an mtDNA construct can be designed to spontaneously form a double stranded hairpin structure containing a transcriptionally active dsDNA T7 promoter sequence when the plasmid is transferred to the mitochondria by conjugation (Yoon & Koob, 2005). The fluorescent protein expression from this construct, therefore, is a direct indicator of transfer of the DNA construct into the mitochondrial matrix and is not dependent on uncharacterized secondary events, such as DNA replication. In addition, because the conjugative process is fairly nonspecific, we could increase the specificity of this reaction for mitochondrial transfer by expressing peptides on the surface of the E. coli (Becker et al., 2005) that are known to bind to the mitochondrial outer membrane (e.g., Mfn1) (Legros et al., 2002). Because mitofusin is required for the initial tethering between adjacent mitochondria before mitochondrial fusion, the protein would facilitate E. coli to search and attach to the mitochondrial membrane in the cytoplasm.
Perspectives
MtDNA mutations and subsequent mitochondrial dysfunction in cells are clearly related with a wide range of inherited human diseases. Although discovering mtDNA mutations that are involved in clinically devastating diseases has advanced significantly, there are currently little or no available therapies to treat mtDNA diseases due to the lack of technology for introducing modified mitochondrial genomes into the mitochondria of living cells. The current strategy for mitochondrial genome therapies is mainly focused on selectively inhibiting mutant mtDNA using antisense molecules or a restriction enzyme and thus can modulate mutant mtDNA levels in cells. Deliverying of circular DNA constructs, however, would provide a new and invaluable tool to genetically engineer mitochondrial genomes in mammalian cells, which could provide further understanding of mitochondrial function and treatment of human mitochondrial diseases.



 XML Download
XML Download