

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Nitric Oxide (NO) is not only a neurotransmitter but also a neuromodulator which exerts many functions in the nervous system (Esplugues, 2002; Rhyu et al., 2003; Abbott & Nahm, 2004). Since NO can cross the lipid bilayer freely and has a very short lifespan, neurons cannot sequester NO nor regulate its local concentration (Dawson & Dawson, 1996). Therefore, the key to regulating NO in the brain is to control NO synthesis by regulating the activity of neuronal NO synthase (nNOS). This regulation of NO synthesis is mainly mediated by cytosolic Ca2+ levels. The Ca2+ influx from extracellular fluid and the release of Ca2+ from intracellular stores increase Ca2+ concentrations in the neuronal cytoplasm. Increased Ca2+ binds calmodulin (CaM) and then the Ca2+CaM complex activates nNOS by direct binding. If the Ca2+ concentration falls, it dissociates from CaM, which in turn dissociates from nNOS resulting in nNOS deactivation (Knowles et al., 1989; Sheng et al., 1992).
While the synthesis of NO is regulated by Ca2+, NO can also influence Ca2+ levels in neuronal cytoplasm. NO diminishes activity of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) (Lei et al., 2000) and N-methyl-D-aspartate (NMDA)-type glutamate receptor (Lei et al., 1992; Manzoni et al., 1992). NO inhibits voltage-gated Ca22+channels such as L-type (Doerner & Alger, 1988) and N-type Ca2+ channels (Yoshimura et al., 2001). The increase of Ca2+ concentration through these receptors and channels can be reduced by these means. Not only Ca2+ influx from extracellular fluid but also Ca2+ release from intracellular Ca2+ stores are modulated by NO. NO induces ryanodine receptor phosphorylation through protein kinase G, which results in increased Ca2+ release from the endoplasmic reticulum into the cytoplasm (Clementi et al., 1996). Therefore it can be said that NO actively participates in the regulation of Ca2+ homeostasis of neurons.
The entire neuronal Ca2+ homeostasis regulation system consists of a Ca2+ entry system, intracellular Ca2+ store, Ca2+ extrusion system, and Ca2+ buffer. It can be hypothesized that NO participates in the regulation of Ca2+ homeostasis through mechanisms other than modulating the Ca2+ entry system and intracellular Ca2+ store. Previously it was shown that Ca2+ binding proteins (CaBPs) such as calbindin-D28k (CB) (Geula et al., 1993; Bertini et al., 1996) and calretinin (CR) (Arévalo et al., 1993) colocalize with nNOS in some populations of neurons. Similar cerebellar function defects are detected in both nNOS (Nelson et al., 1995) and CaBP knock-out mice (Airaksinen et al., 1997; Cheron et al., 2000). Based upon these findings, Ca2+ buffer may be a candidate for Ca2+ homeostasis regulation by NO. It is well known that CaBPs such as CB, CR, and parvalbumin (PV) act as Ca2+ buffers in neurons (Schwaller et al., 2002) and that nNOS and these proteins are abundantly expressed and exert several functions in the cerebellum (Nelson et al., 1995; Schwaller et al., 2002). Therefore, to test NO's influences on these Ca2+ buffer proteins, we examined changes in their expression in the cerebellum of nNOS knock-out mice (nNOS(-/-) mice) (Huang et al., 1993) using immunohistochemistry. We were able to demonstrate specific changes in expression of each Ca2+ buffer protein in the cerebellum of the nNOS(-/-) mice.
Materials and Methods
Male mice 3~4 months old were utilized for this study. There were 12 C57BL/6 controls and 10 nNOS(-/-) B6, 129S-Nos1tm1Pih obtained from Dr. Oh (Induced Mutant Resources Program, Genetic Resources Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, Korea). All animals were treated in accordance with the 'Principles of Laboratory Animal Care' (NIH publication No. 86~23, revised in 1985). The mice were perfused transcardially with cold phosphate buffered saline (PBS, 0.05M, pH 7.4), followed by ice-cold 4% paraformaldehyde. The brains were cryoprotected in a series of cold sucrose solutions, and cut at 40 µm in the coronal plane. Immunohistochemistry was performed in accordance with the free-floating method described earlier (Chung et al., 2000). Rabbit anti-CB polyclonal antibody, rabbit anti-CR polyclonal antibody, and anti-PV monoclonal antibody (AB1778 and AB5054; Chemicon International, Temecula, CA, USA for CB and CR respectively and P3088; Sigma, Saint Louis, MI, USA for PV) were used as primary antibodies.
A sample of sections was reacted without primary antiserum, and different samples were exposed to primary antiserum that had been preabsorbed for 24 hours with control antigen peptides. Sections from these samples did not exhibit any immunoreactivity as described in this report (Fig. 1). We randomly selected 5 unit areas at each region in the cerebellum from control (n=12) and nNOS(-/-) mice (n=10) and calculated the numbers of CB, CR, and PV-ir per unit area. Sections from each control and nNOS(-/-) group were stained together eliminating the variable of different experimental conditions. Visual assessment and densitometric measurements using an NIH image program (Scion Image) were evaluated to determine staining density. The t-test was used to investigate whether changes in CaBPs expression were statistically significant (*P<0.01).
Immunohistochemistry images were taken using a ProgRes C14 digital camera with ProgRes C14 software (JENOPTIC Laser, Optic, System, Munich, Germany). Image-editing software (Adobe Photoshop) was used to adjust size and contrast and combine the images obtained.
Results
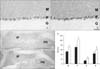
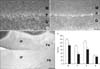
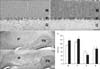
In the cerebellar cortex, each CaBP showed a characteristic expression pattern. CB-immunoreactivity was observed in the neuropil of the molecular layer and cell bodies of Purkinje cells (Fig. 2A) whereas CR-immunoreactivity was confined to neuropil of the molecular layer and granular cells of the granular layer (Fig. 3A). PV was expressed by the neuropil, cells of the molecular layer, and cell bodies of Purkinje cells (Fig. 4A). These expression patterns of CB, CR, and PV were well preserved in the nNOS(-/-) mice (Fig. 2A, B for CB; Fig. 3A, B for CR; Fig. 4A, B for PV). However, the immunoreactivity of each CaBP was significantly changed. In all layers of the cerebellar cortex, CB-immunoreactivity was significantly increased (Fig. 2A, B, E) and in contrast to CB, CR-immunoreactivity was significantly decreased (Fig. 3A, B, E). Regarding PV, PV-immunoreactivity of the molecular layer and the Purkinje cell layer were significantly increased whereas that of the granular layer was significantly decreased (Fig. 4A, B, E).
Immunoreactivity of CB and CR in the deep cerebellar nuclei was much lower than that in the cerebellar cortex and most were confined in neuropil (Fig. 2C, 3C). Similar to changes in the cerebellar cortex, CB-immunoreactivity of the nNOS(-/-) mice was significantly increased (Fig. 2D, E) and CR-immunoreactivity of the nNOS(-/-) mice was decreased, although this change was not statistically significant (Fig. 3D, E). In contrast to CB and CR, relatively high levels of PV were observed in the deep cerebellar nuclei and expressed not only in neuropil but also in cell bodies (Fig. 4C). Immunoreactivity was similar between control and nNOS(-/-) groups (Fig. 4C, D, E) but cell bodies of nNOS(-/-) mice appeared to express higher PV. (Fig. 4C, D).
Previously the distribution of CB, CR, and PV in the cerebellum was carefully examined and their characteristic localizations reported (Celio, 1990; Résibois & Rogers, 1992). In the molecular layer, stellate cells and basket cells expressed PV, parallel fibers of granule cells expressed CR, and dendrites of Purkinje cells expressed CB and PV. In the Purkinje cell layer the cell bodies of Purkinje cells showed CB and PV-immunoreactivity. In the granular layer only CR was expressed by granular cells. These characteristic expressions of CB, CR, and PV match well with our results (Fig. 2~4). Therefore CB and CR-immunoreactivity in neuropil of the molecular layer are thought to be due to dendrites of Purkinje cells and parallel fibers of granule cells respectively. PV-immunoreactivity of cells and neuropil of the molecular layer may be due to stellate cells, basket cells, and dendrites of Purkinje cells.
The CB, CR, and PV actively participate in the regulation of neuronal intracellular Ca2+ levels by binding Ca2+ and lowering its concentration. The intracellular steady state Ca2+ level of neurons is not affected by these CaBPs (Chard et al., 1993; Schwaller et al., 2002), because it is determined by Ca2+ uptake and extrusion system which remains functional until Ca2+ has attained its steady state level. Instead, these calcium buffer proteins modulate the temporal and spatial properties of intracellular Ca2+ distribution. For example, CB significantly reduces the amplitude of the Ca2+ transient and slows down the decay of Ca2+ levels in cerebellar Purkinje cells (Airaksinen et al., 1997). PV, which has slower Ca2+ binding properties than CB, increases the initial rate of decay of Ca2+ and subsequently prolongs its late phase in bovine chromaffin cells (Lee et al., 2000). Therefore, it is possible that NO influences intracellular Ca2+ levels of the cerebellar neurons by modulating CaBPs expression levels.
Discussion
The nNOS(-/-) mice have no evident defects in usual locomotor activity (Huang et al., 1993) but show discrete abnormalities in balance and motor coordination selectively at night (Kriegsfeld et al., 1999). Similarly, CB knock-out mice (Airaksinen et al., 1997) and Purkinje cell-specific conditional CB knock-out mice (Barski et al., 2000) show normal motor functions in usual environments but slip when transversing narrow runways, which force them to change and adapt their stride. Also in CR knock-out mice, abnormal Purkinje cell activity in the cerebellum and abnormal wheel running test results are observed whereas other motor functions remain normal (Schanne et al., 1979; Cheron et al., 2000). Regarding these findings, the nNOS(-/-) mice, the CB knock-out mice, and the CR knock-out mice seem to have very similar cerebellar functional defects, although motor function tests used are not exactly the same. This suggests that there may be a common underlying mechanism. Since altered CB and CR expression in the nNOS(-/-) mice was observed in this study, altered expression of these proteins and resulting temporal and spatial intracellular Ca2+ distribution property changes may be good candidates for that common mechanism.
It is well known that intracellular concentration of Ca2+ and the Ca2+-dependent signaling system including NOS are closely related to neuronal degenerations (Schanne et al., 1979; Lipton et al., 1993). The Ca2+ buffering property of CB, CR, and PV allows for the hypothesis that these buffers may have neuroprotective effects. There is some in vitro data supporting this hypothesis (D'Orlando et al., 2001; D'Orlando et al., 2002). In spinocerebellar ataxia type I, it is suggested that down-regulation of CB and PV leads to cerebellar Purkinje cell death (Vig et al., 2000). Many studies have also shown results against this hypothesis (Kuźnicki et al., 1996; Airaksinen et al., 1997; Bouilleret et al., 2000). In CB, CR, and PV knock-out mice there is no evidence of abnormal neuronal loss (Schwaller et al., 2002), resulting in doubts as to a generalized neuroprotective role for these proteins. Like the Ca2+ buffer proteins, no unusual neuronal death is observed in the nNOS(-/-) mice (Huang et al., 1993; Nelson et al., 1995); therefore, it is not obvious that Ca2+ buffer proteins such as CB, CR, and PV have generalized neuroprotective effects. Their modulated expression by NO seems to have minor effects on neuronal survival.
In the present study, we demonstrated that expression levels of CB, CR, and PV are characteristically and significantly altered in the cerebellum of nNOS(-/-) mice using immunohistochemistry techniques (Figs. 2~4). These changing patterns were preserved in all animals studied. Regarding these findings, it can be concluded that NO modulates CaBP expression in neurons of the cerebellum, and by this means, NO participates in Ca2+ homeostasis and regulation of neurons. As expression patterns of these proteins are not changed, it appears that NO influences only the expression level, not the expression pattern in the cerebellum.
For the first time, we demonstrated that NO specifically modulates the expression of Ca2+ buffer proteins such as CB, CR, and PV in the cerebellum. This result suggests another mechanism by which NO participates in the regulation of Ca2+ homeostasis. Since modulation of expression levels of Ca2+ buffer proteins can influences temporal and spatial properties of intracellular Ca2+ distribution, it appears that NO can exerts its various functions not only in the cerebellum but also in the other parts of the brain. The exact mechanism of this regulation and its functional significance requires further elucidation.

 XML Download
XML Download