

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Stress, a nonspecific response of the body, has long been believed to be associated with various forms of diseases (Selye, 1980). In the brain, the hippocampus is sensitive to stress and is one of the first regions to be structurally and functionally modified by severe and inescapable stress (McEwen, 2000), because the hippocampus is one of the target brain regions hippocampusof corticosterone. In animal and human studies stress significantly decreases dendritic length. The number of branch points in hippocampal CA3 pyramidal neurons induces atrophy of the apical dendrites of CA3 pyramidal cells in the rat hippocampus, and suppresses hippocampal learning and memory (Watanabe et al., 1992; Magariños & McEwen, 1995; Magariños et al., 1996; Vyas et al., 2002).
Transient occlusion of the common carotid artery deprives the brain of blood flow and significantly reduces glucose and oxygen levels. This condition induces neuronal changes in the central nervous system. Among susceptible regions, the hippocampus is the most vulnerable region to cerebral ischemia, and extensive neuronal death occurs in this region (Kirino, 1982; Petito & Pulsinelli, 1984). Recent studies have identified inflammation as a possible mechanism in neuronal damage induced by cerebral ischemia because inflammatory cells are known to infiltrate the ischemic brain area (Gautier et al., 2009; Hendryk et al., 2010). In addition, some studies have shown that stress leads to an exacerbation of stroke outcomes (DeVries et al., 2001: Sugo et al., 2002). In contrast, others reported that stress reduced brain damage after an ischemic insult (Thoresen et al., 1996).
Platelet endothelial cell adhesion molecule-1 (PECAM-1, CD31) is a very important mediator in inflammation; it contributes to leukocyte transendothelial migration during inflammation (Muller & Randolph, 1999; O'Brien et al., 2003). PECAM-1 levels increase significantly in serum and cerebrospinal fluid of patients within 24 hours after an ischemic stroke (Zaremba & Losy, 2002b). In addition, PECAM-1 immunoreactivity and protein levels have been shown to be significantly increased in the hippocampal CA1 region 4 days after ischemia/reperfusion (Hwang et al., 2005). The elevation of PECAM-1 in vessels is known to increase the transmigration of lymphocytes and neutrophils (Dangerfield et al., 2002; Michiels et al., 1998). Infiltrative neutrophils and monocytes produce neural toxic substances and contribute to neuronal damage following ischemic stroke (Wang et al., 2002; Deng et al., 2003; Gidday et al., 2005). In addition, inflammation by infiltration of activated neutrophils and microglia is known as a major contributor of secondary neuronal death after a stroke (Iadecola & Alexander, 2001; Zheng et al., 2003; Williams et al., 2004). However, to date there are no studies documenting the relationship between stress and PECAM-1 expression in the brain after an ischemic insult. Therefore, in the present study, we investigated the effects of stress on ischemic damage, and PECAM-1 immunoreactivity and protein levels in the gerbil hippocampus following 5 minutes of transient cerebral ischemia.
Materials and Methods
Experimental animals
The progeny of male Mongolian gerbils (Meriones unguiculatus)
were obtained from the Experimental Animal Center, Hallym University, Chuncheon, South Korea. Gerbils were used at 6 months (B.W., 65~75 g) of age. The animals were housed in a conventional state under adequate temperature (23℃) and humidity (60%) control with a 12-h light/12-h dark cycle, and provided with free access to water and food. The procedures for handling animals and their care conformed to the guidelines that are in compliance with current international laws and policies (NIH Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85~23, 1985, revised 1996) and were approved by the Hallym's Medical Center Institutional Animal Care and Use Committee (IACUC). All of the experiments were conducted to minimize the number of animals used and suffering caused by such procedures.
Induction of repeated restraint stress
The restraint stress cages were consisted of adjustable length (12.0~2.0 cm length and 3.8 cm diameter) Plexi-glass tubes with air holes in the front, top and back. Gerbils were divided into two groups; control- and repeated restraint stress (stress)-groups. Control-group was left untouched in their home cages until the ischemic surgery. Stress-group was placed into restrainers for 5 hours (between 09:30 h and 14:30 h) for 21 consecutive days prior to the ischemic surgery. Finally, experimental groups were divided into 4 groups; 1) sham-operated control-group (sham-group), 2) ischemia-operated control-group (ischemia-group), 3) sham-operated stress-group (stress-sham-group), 4) ischemia-operated stress-group (stress-ischemia-group).
Induction of transient cerebral ischemia
The animals in both control and stress groups were anesthetized with a mixture of 2.5% isoflurane (Baxtor, Deerfield, IL) in 33% oxygen and 67% nitrous oxide. Bilateral common carotid arteries were isolated and occluded using non-traumatic aneurysm clips. The complete interruption of blood flow was confirmed by observing the central artery in retinae using an ophthalmoscope. After 5 minutes of occlusion, the aneurysm clips were removed from the common carotid arteries. The body (rectal) temperature under free-regulating or normothermic (37±0.5℃) conditions was monitored with a rectal temperature probe (TR-100; Fine Science Tools, Foster City, CA) and maintained using a thermometric blanket before, during and after the surgery until the animals completely recovered from anesthesia. Thereafter, animals were kept on the thermal incubator (Mirae Medical Industry, Seoul, South Korea) to maintain the body temperature of animals until the animals were euthanized. Sham-operated animals were subjected to the same surgical procedures except that the common carotid arteries were not occluded.
Tissue processing for histology
For the histological analysis, animals were anesthetized with sodium pentobarbital (30 mg/kg, i.p.) and perfused transcardially with 0.1 M phosphate-buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde in 0.1 M phosphate-buffer (PB, pH 7.4). The brains were removed and postfixed in the same fixative for 6 hours. The brain tissues were cryoprotected by infiltration with 30% sucrose overnight. Thereafter, frozen tissues were serially sectioned on a cryostat (Leica, Wetzlar, Germany) into 30-µm coronal sections, and they were then collected into six-well plates containing PBS.
Cresyl violet staining
To examine the delayed neuronal death in the hippocampal CA1 region after transient cerebral ischemia, cresyl violet staining was performed. The sections were mounted on gelatin coated microscopic slides. Cresyl violet acetate (Sigma, St. Louis, MO) was dissolved at 1.0% (w/v) in distilled water, and glacial acetic acid was added in this solution. The sections were stained and dehydrated by immersing in serial ethanol baths, and they were then mounted with Canada balsam (Kanto, Tokyo, Japan).
Immunohistochemical staining for PECAM-1
To obtain the accurate data for PECAM-1 immunoreactivity,
the sections from sham- and ischemia-operated animals in both control and stress groups (n=7 at each time point) were used at designated times (1, 2 and 4 days) after the ischemic surgery under the same conditions. The sections were sequentially treated with 0.3% hydrogen peroxide (H2O2) in PBS for 30 minutes and 10% normal goat serum in 0.05 M PBS for 30 minutes. They were next incubated with diluted mouse anti-PECAM-1 antibody (diluted 1 : 1,000, sc-1506, Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4℃ and subsequently exposed to biotinylated goat anti-mouse IgG and streptavidin peroxidase complex (diluted 1 : 200, Vector, Burlingame, CA). And they were visualized by staining with 3,3'-diaminobenzidine (Sigma, St. Louis, MO) in 0.1 M Tris-HCl buffer (pH 7.2) and mounted on gelatin-coated slides. After dehydration the sections were mounted with Canada balsam (Kanto, Tokyo, Japan). A negative control test was carried out using pre-immune serum instead of primary antibody in order to establish the specificity of the immunostaining. The negative control resulted in the absence of immunoreactivity in all structures.
All measurements were performed in order to ensure objectivity in blind conditions, by two observers for each experiment, carrying out the measures of control and experimental samples under the same conditions.
Ten sections per animal were serially selected from the corresponding areas of the hippocampus in order to quantitatively analyze PECAM-1 immunoreactivity in this region. The studied tissue sections were selected according to anatomical landmarks corresponding to AP -1.4~-1.8 mm of gerbil brain atlas (Loskota et al., 1974). The corresponding areas of the hippocampus were measured on the monitor at a magnification of 25~50×. Images of all PECAM-1 immunoreactive structures, taken from 3 layers (strata oriens, pyramidale and radiatum in the hippocampal CA1-3 regions and polymorphic, molecular and granule cell layer in the dentate gyrus), were obtained through an AxioM1 light microscope (Carl Zeiss, Göttingen, Germany) equipped with a digital camera (Axiocam, Carl Zeiss, Göttingen, Germany) connected to a PC monitor. The immunostaining density of all PECAM-1 immunoreactive structures was evaluated on the basis of a optical density (OD), which was obtained after the transformation of the mean gray level using the formula: OD=log (256/mean gray level). The OD of background was taken from areas adjacent to the measured area. After the background density was subtracted, a ratio of the optical density of image file was calibrated as % (relative optical density, ROD) using Adobe Photoshop version 8.0 and then analyzed using NIH Image 1.59 software.
Western blot analysis for PECAM-1
To confirm changes in PECAM-1 levels in the hippocampus following transient cerebral ischemia, sham- and ischemia-operated animals in both control and stress groups (n=7 at each time point) were used for western blot analysis at 4 days after ischemia/reperfusion. After sacrificing animals, the hippocampus was removed. The tissues were homogenized in 50 mM PBS (pH 7.4) containing 0.1 mM ethylene glycol bis (2-aminoethyl Ether)-N,N,N',N' tetraacetic acid (EGTA) (pH 8.0), 0.2% Nonidet P-40, 10 mM ethylendiamine tetraacetic acid (EDTA) (pH 8.0), 15 mM sodium pyrophosphate, 100 mM β-glycerophosphate, 50 mM NaF, 150 mM NaCl, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1 mM dithiothreitol (DTT). After centrifugation, the protein level was determined in the supernatants using a Micro BCA protein assay kit with bovine serum albumin as the standard (Pierce Chemical, Rockford, IL). Aliquots containing 20 µg of total protein were boiled in loading buffer containing 150mM Tris (pH 6.8), 3 mM DTT, 6% SDS, 0.3% bromophenol blue and 30% glycerol. The aliquots were then loaded onto a 10% polyacrylamide gel. After electrophoresis, the gels were transferred to nitrocellulose transfer membranes (Pall Crop, East Hills, NY). To reduce background staining, the membranes were incubated with 5% non-fat dry milk in PBS containing 0.1% Tween 20 for 45 minutes, followed by incubation with mouse anti-PECAM-1 antiserum (1 : 2,000), peroxidase-conjugated goat anti-mouse IgG (Sigma, St. Louis, MO) and an ECL kit (Pierce Chemical, Rockford, IL).
The result of the western blot analysis was scanned, and densitometric analysis for the quantification of the bands was done using Scion Image software (Scion Corp., Frederick, MD), which was used to count relative optical density (ROD): A ratio of the ROD was calibrated as %, with sham-group designated as 100%.
Statistical analysis
The data shown here represent the means±SEM. Differences among the means were statistically analyzed by two-tailed Student t-test in order to elucidate the effects of repeated restraint stress against ischemic damage. In addition, differences of the mean ROD among the groups were statistically analyzed by analysis of variance (ANOVA) followed by Tukey's multiple range method in order to elucidate ischemia-related differences among experimental groups. Statistical significance was considered at p<0.05.
Results
Neuronal damage
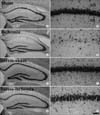
The effect of repeated restraint stress on delayed neuronal death in the hippocampal CA1 region 4 days after ischemia/reperfusion was examined by cresyl violet staining. In the sham- and stress-sham groups, cresyl violet (CV) positive (+) cells were distributed normally in the subregions of the gerbil hippocampal formation including the hippocampal CA1 region (Fig. 1A, B, E, F, 2). Four days after ischemia/reperfusion, in the ischemia-group, only a few of CV+ cells were detected in the stratum pyramidale of the hippocampal CA1 region due to the delayed neuronal death; however, we could not find significant neuronal damage in the other subregions such as the CA2/3 region and dentate gyrus (Fig. 1C, D, 2). In the stress-ischemia-group, most of neurons in the hippocampal formation were stained with CV (Fig. 1G, H); about 82.2% of pyramidal neurons in the hippocampal CA1 region were stained with CV (Fig. 2).
Changes in PECAM-1 immunoreactivity
In the sham- and stress-sham-groups, PECAM-1 immunoreaction
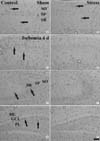
was detected in blood vessels, located mainly in the stratum oriens and radiatum of the CA1-3 region (Fig. 3A, B) and in the molecular and polymorphic layers of the dentate gyrus (data not shown). In both the ischemia- and stress-ischemia-groups, PECAM-1 immunoreactivity was not changed significantly until 2 days after ischemia/reperfusion (data not shown). Four days after ischemia/reperfusion, PECAM-1 immunoreactivity was significantly increased in the ischemia-groups (Fig. 3C, E, G, 4). However, PECAM-1 immunoreactivity in the CA2/3 region and dentate gyrus was lower than that in the CA1 region (Fig. 4). At this time point after ischemia/reperfusion, PECAM-1 immunoreactivity in the stress-ischemia-group was much lower than that in the ischemia-group, and was similar to that in the sham-groups (Fig. 3D, F, H, 4).
Changes in PECAM-1 protein levels
In the western blot study, we found that changes in PECAM-1 protein levels in the gerbil hippocampus after ischemia/reperfusion were generally similar to the immunohistochemical change. PECAM-1 protein levels were similar between the sham- and stress-sham-groups. Four days after ischemia/reperfusion, PECAM-1 protein level was significantly increased in the ischemia-group: PECAM-1 protein level was increased to about 340% of that in the sham-group. However, there was no significant increase on PECAM-1 protein level in the stress-ischemia-group: it was similar to that in the sham-group (Fig. 5).
Discussion
In this study, we observed that repeated restraint stress protected ischemia-related neuronal death in the hippocampal
CA1 region after ischemia/reperfusion. Previous studies showed the relationship between stress and neuronal damage following ischemic insults. In a permanent focal cerebral ischemic rat model of subacute immobilization stress (1 hour for 7 days), infarct size increased and behavioral scores decreased, whereas chronic stress (6 hours for 21 days) decreased infarct size (Madrigal et al., 2003). It is also known that repeated short-term stress (45 minutes for 7days) induces vulnerability to neuronal damage following middle cerebral artery occlusion (Sugo et al., 2002). It has also been reported that repeated stress increases the vulnerability of hippocampal cells in an in vitro model of ischemia (Fontella et al., 2005). Although there are some discrepancies, our results are supported by the findings of previous studies which showed that repeated restraint stress protected brain damage induced by an ischemic insult. Additionally, it is well known that repeated same stress can lead to a process of adaptation to stress-induced changes (Martí et al., 1993; Lachuer et al., 1994; Cancela et al., 1995).
In the present study, we observed the changes of PECAM-1 immunoreactivity and protein levels in the hippocampus of control- and stress-groups after transient cerebral ischemia. In the ischemia-group, both PECAM-1 immunoreactivity and protein levels were significantly increased in all the hippocampal subregions 4 days after ischemia/reperfusion. This result coincided with a previous study which reported that PECAM-1 immunoreactivity and protein levels were significantly increased in the hippocampal CA1 region 4 days after ischemia/reperfusion (Hwang et al., 2005). This may be associated with neutrophil transmigration because PECAM-1 plays an important role in the adhesion cascade leading to leukocyte-endothelial transmigration and extravasation of PECAM-1 producing cells, such as neutrophils, monocytes, and lymphocytes during inflammation (Ilan & Madri, 2003; Kalinowska & Losy, 2006). It has also been reported that serum PECAM-1 levels were significantly elevated in stroke patients (Zaremba & Losy, 2002a, b). In addition, we observed a considerable increase of PECAM-1 immunoreactivity in the CA1 region of the ischemia-group, compared to a moderate increase in the CA2/3 region and dentate gyrus. Kirino (1982) reported that different types of neuronal damage were observed in the CA1 region, CA2/3 region, and dentate gyrus after transient cerebral ischemia in gerbils. The changes in the CA1, CA2/3, and dentate gyrus were termed delayed neuronal death, reactive change, and ischemic cell change, respectively. The degree of increase in PECAM-1 may therefore be associated with the regional vulnerability post transient cerebral ischemia.
However, in this study, we observed that PECAM-1 immunoreactivity
and protein levels did not change in the hippocampus
of the stress-ischemia-group. It was reported that restraint stress, in itself, did not induce inflammatory cytokines
in the periphery or brain (Maier et al., 1999; Deak et al., 2003, 2005; Sugama et al., 2007). In addition, it has been shown in a rat myocardial infarction model that blocking PECAM-1 exerts a significant protective effect via blockade of neutrophil accumulation in the myocardium, and that anti-PECAM-1 antibodies suppress neutrophil accumulation into the inflamed peritoneum of rats (Vaporciyan et al., 1993; Gumina et al., 1996). PECAM-1 antibodies also block transendothelial migration of leukocytes (Muller et al., 1993; Muller, 1995) and reduce the platelet adhesion/aggregation in vivo at the site of endothelial injury in brain arterioles (Rosenblum et al., 1994). Moreover, PECAM-1-deficient mice are viable but exhibit defects in angiogenesis and inflammation (Duncan et al., 1999; Thompson et al., 2001; Graesser et al., 2002; Solowiej et al., 2003; Schenkel et al., 2004). As mentioned previously, inflammation and infiltration of inflammatory cells are thought to be important factors in ischemia-induced neuronal damage (Gautier et al., 2009; Hendryk et al., 2010); therefore, the fact that there were no significant changes in PECAM-1 immunoreactivity and protein levels in the stress-ischemia-group may be related to the blockade of inflammatory events such as infiltration of inflammatory cells.
In conclusion, repeated restraint stress protected transient ischemia-induced neuronal death and maintained PECAM-1 immunoreactivity and protein levels in the hippocampus following ischemia/reperfusion. These results indicate that the fact that we detected no significant changes in PECAM-1 may be connected to the effects of repeated restraint stress and ischemic damage.



 XML Download
XML Download