

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Stroke is a leading cause of death and disability in many industrial nations, yet treatments are few and often restricted to few of its victims. Research into identifying important therapeutic targets has led to the realization that the inflammatory reaction accompanying acute stroke appears to play a largely detrimental role in stroke outcome (Wang et al., 2007). Thus, the identification of appropriate anti-inflammatory treatments and understanding their mechanism would be of translational importance. Microglia are the brain's resident immune cell, and are thought to be immune cells of myeloid origin. While they play important roles in brain development and recovery from various neurological injuries including stroke, their early activation may cause worsening of ischemic injury. In fact, several studies suggest that under some settings, microglia tend to worsen neuronal damage (Flavin et al., 1997; Flavin & Ho, 1999; Flavin & Zhao, 2001; Flavin et al., 2000; Siao et al., 2003). In order to explore the possibility that microglia may be an important early therapeutic target, we use a cell culture model of ischemia-like injury to understand the contribution of microglia to such injury, and whether inhibiting its activation may be therapeutic.
Microglia generate a variety of toxic substances which may exacerbate ischemic cell death (Wang et al., 2007). We previously showed that microglia potentiate injury to co-cultures of astrocytes and endothelial cells subjected to ischemia-like insults (Yenari et al., 2006; Zheng et al., 2008). Minocycline, a tetracycline family antibiotic, is recognized to have anti-inflammatory properties, and as such is known to protect against brain ischemia through this mechanism (Yrjänheikki et al., 1998; Yrjänheikki et al., 1999; Tang et al., 2007). We previously showed that minocycline, by preventing microglial activation, could prevent potentiation of ischemia-like injury to astrocytes and endothelial cells (Yenari et al., 2006). Here, we explore the extent to which microglia may be involved in potentiating neuronal death in an in vitro model of ischemia-like injury, and whether anti-inflammatory treatments only protect in the presence of added microglia, or whether direct effects on neurons can be demonstrated.
Materials and Methods
Cell culture & ischemia-like injury
After obtaining approval from institutional committees on laboratory animal use and care, primary neuron (N) and microglial cultures (M) were prepared from Swiss Webster mice using methods previously described (Dugan et al., 1995; Yenari & Giffard, 2001). All experiments were carried out in accordance with the NIH Guide for the Care and Use of Laboratory animals (NIH Pub. 80-23). Measures were taken to minimize pain and discomfort. Both N and M cultures contain <5% astrocytes based on immunolabeling with GFAP. Cocultures of neurons and microglia (NM) were prepared by seeding 2×105 microglia/ml (M) onto neuron cultures (N), and allowing the cocultures to stabilize 24 h prior to use in experiments (Fig. 1). The cultures (N, M, and NM) were treated with minocycline (2 µM) to inhibit microglial activation or vehicle. Alternatively, cultures were treated with aminoguanidine (200 µM, Sigma, St. Louis, MO) to inhibit inducible nitric oxide synthase (iNOS), GM6001 (20 µM, Chemicon, Temecula, CA) a broad spectrum matrix metalloprotease (MMP) inhibitor, a TNF-alpha blocking antibody (1 µg/ml, #AF-410-NA, R&D Systems, Minneapolis, MN) or plasminogen activator inhibitor-1 (PAI-1, 1 µg/ml, American Diagnostica Inc., Stamford. CT) an endogenous inhibitor of tissue plasminogen activator (tPA). The doses of reagents used were based on prior literature reports and manufacturer's suggestions.
Cell cultures were subjected to ischemia-like injury through oxygen and glucose deprivation (OGD) for 90 min by placing cultures in an anaerobic chamber (O2<0.2%, Coy Laboratories, East Lansing, MI) in a glucose-free balanced salt solution (BSS0). After 90 min of OGD, cultures were "reperfused" by adding 5.5 mM glucose to the media at normoxia. Control cultures (no injury) were incubated with balanced salt solution containing 5.5 mM glucose (BSS5.5). Inhibitors were added to the final BSS0 wash after the cells were put in the anaerobic chamber.
Cultures were also treated with lipopolysaccharide (LPS, E. Coli serotype 055:b5, Sigma) as previously described by us (Yenari & Giffard, 2001). (LPS, E. Coli serotype 055:B5; Sigma). A concentration of 10 µg/ml was applied by diluting in serum-free media. This concentration was chosen as it was an intermediate concentration studied by our group, and not found to cause toxicity to primary microglia. It also led to consistent activation as manifested by transformation into amoeboid morphology and secretion of nitric oxide(Yenari & Giffard, 2001; Han et al., 2002). Cultures were washed three times in LPS containing media, then returned to the incubator.
Assessment of cell death
Cell death was assessed by trypan blue staining. Cells with neuronal morphology were identified and counted in a blinded fashion. Ten random high power fields were selected, and total cell counts and counts of trypan blue positive (dead) cells were made. Cells were only counted if they exhibited typical neuronal morphology (large, rounded cell bodies with long processes, which distinguish them from astrocytes and microglia (Ying et al., 1999)).
A second biochemical assay was also used, based on release of lactate dehydrogenase (LDH) released in to the media, and expressed as a percent of the total LDH releast after freeze-thaw (=100%) (Lee et al., 2001).
Experiments were performed in triplicate from 3 separate dissections.
Assays
Nitric oxide (NO) levels in culture media was estimated using the Griess reagent (Sigma) using previously published methods (Han et al., 2002), and TNF-α levels were measured by ELISA (OptEIA, BD Biosciences, San Jose, CA) following kit instructions.
Immunostaining was performed in cells following fixation in acetone precooled to -20C for 10 minutes at 4C, then rinsed in PBS, and blocked in normal serum. Primary antibody against MAP-2 (1 : 200, Sigma) to identify neurons was applied followed by a Texas-red labeled secondary antibody (1 : 200, Jackson Labs). Between steps, sections were washed 3 times in PBS. Microglia were identified by staining with FITC-labeled Griffonia simplicifolia isolectin B4 (IB4, 10 µg/ml, Sigma) (Han et al., 2002).
Results
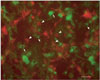
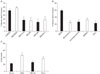
Immunostaining of neuron-microglia co-cultures is shown in Fig. 1. Neurons are identified by MAP-2 staining in red, as angulated cells with long slender processes. Microglia, shown in green by isolectin B4 (IB4), appear to have compact cell bodies with shorter processes (resting form), and no processes with larger, rounded cell bodies in the activated form. Microglia were added to neuronal cultures for OGD experiments on NM co-cultures. Microglia increased the extent of cell death to a small extent compared to neurons alone (Fig. 2A). Thus, microglia appear to be somewhat toxic to neurons following OGD.
Treatment of NM cocultures with minocycline at the time of OGD led to a marked reduction in cell death, even beyond that due to the addition of microglia (Fig. 2A). Similarly, applying inhibitors of several inflammatory mediators led to similar amounts of protection. Interestingly, the extent of neuroprotection due to minocycline as well as all the other inhibitors tested was greater than that due to microglia-induced toxicity, suggesting that these treatments might have a direct effect on neurons. This would suggest that while microglia may be responsible for some increased neuron death, neurons themselves may produce similar inflammatory factors that could lead to their own death. Next, we treated relatively pure neuronal cultures with minocycline or the above inhibitors. In all cases, treatment reduced OGD-induced injury in neuronal cultures suggesting that inhibiting factors traditionally thought to emanate from immune cells can directly protect neurons without added microglia (Fig. 2B). Similar observations were made when neuron cultures were treated with a blocking antibody against TNF-alpha (Fig 2C).
To establish that minocycline has anti-inflammatory effects in our hands, primary microglia or BV2 cells (a cell line of transformed murine microglia (Blasi et al., 1990)) were subjected to 24 h OGD followed by 2 h reperfusion or stimulation with lipopolysaccharide (1~10 µg/ml LPS, Escherichia coli serotype 055:B5; Sigma, St. Louis, MO) for 24 h. TNF-α and NO levels were measured in culture supernatants. Both LPS and OGD increased TNF-α levels in primary microglia, and this was markedly inhibited by minocycline (Figs. 3A and B). In contrast, LPS, but not OGD increased NO production, and this was not inhibited by minocycline in BV2 cells (Fig. 3C) or primary microglia (Fig. 3D).
To determine whether neurons generate inflammatory mediators, and whether these could be inhibited by minocycline, we measured levels of TNF-α and NO in the media of neuron cultures subjected to OGD. Interestingly, neurons alone generated low levels of TNF-α and NO, but only NO was increased by OGD and suppressed by minocycline (Fig. 4).
Discussion
Microglia somewhat worsened injury to neurons in vitro, and this increased injury was more than blocked by minocycline, an inhibitor of microglia activation (Tikka & Koistinaho, 2001). While these observations are in line with prior reports, as well as recent publications from our labs showing that minocycline is both anti-inflammatory and neuroprotective using in vivo models (Yrjänheikki et al., 1999; Yenari et al., 2006; Tang et al., 2007), it raised the question of whether minocycline has direct effects on neurons. By taking advantage of in vitro models where mechanisms can be studied more precisely, we now show direct protection of neurons alone by minocycline as well as by inhibiting other inflammatory mediators including iNOS, MMPs, TNF-alpha and tPA. This suggests that neurons themselves may be capable of secreting inflammatory mediators largely thought to come from traditional immune cells including microglia, or other glial cells such as astrocytes. In fact, prior reports have documented expression of inflammatory cytokines such as TNF-α in ischemic neurons (Liu et al., 1994) and tPA (Siao et al., 2003), and generation of neuronal tPA has been thought to mediate microglial activation. Protection of neurons alone by the endogenous tPA inhibitor PAI-1 would suggest that neurons are capable of generating substances that are toxic to themselves. Similar conclusions could be made of neuronal generation of iNOS and MMPs. We have previously documented neuronal expression of MMPs (Lee et al., 2001), and a few reports indicate that some neurons can express iNOS after brain ischemia (Corsani et al., 2008). TNF-α has shown conflicting reports in the literature in terms of protection versus potentiation of ischemic cell death (Stoll et al., 2002), but our data here support a damaging effect, as blocking TNF-α clearly reduced neuron cell death following OGD.
Minocycline also exhibited differential inhibition of immune mediators TNF-α and NO which was cell-type and stimulus dependent. While minocycline remarkably inhibited TNF-α secretion in microglia, it did not affect TNF-α secretion in neurons. In contrast, LPS stimulation increased NO generation by microglia, but OGD did not. Minocycline also did not suppress OGD-induced NO generation in microglia, but decreased NO in neurons. Thus, OGD increased TNF-α in microglia, and NO in neurons, while minocycline suppressed these mediators in the respective cell types, and would suggest a complex interplay of immune mediators that cannot be generalized to all cell types.
Some comment on the specificity of minocycline is appropriate here. Originally thought to have anti-microglial properties, beyond its antibiotic activity, minocycline is also known to have anti-apoptotic effects (Zhu et al., 2002; Wang et al., 2003). Tetracycline antibiotics may exert anti-inflammatory effects by multiple mechanisms including inhibiting transcription of the p38 MAPK (Tikka & Koistinaho, 2001) and by cross linking adhesion molecules on the surface of cells (Clark et al., 1994). Regardless, we suggest that our observations argue that further studies for a better understanding of the complexities of immune responses of all cell types in the brain following injury are needed. Understanding inflammatory interactions of all brain cell types is needed to identify how they may be manipulated to provide neuroprotection. Studies of minocycline effects on brain cells in addition to microglia are timely, as minocycline is being studied at the clinical level for various neurodegenerative disorders (Blum et al., 2004; Gordon et al., 2007) and a pilot study suggested efficacy in stroke patients (Lampl et al., 2007).


 XML Download
XML Download