

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Parkinson's disease (PD), the second most common neurodegenerative disease in the world, is characterized by locomotor disorders including rigidity, tremor, bradykinesia, and postural instability (Lang & Lozano, 1998). In addition, massive and selective degeneration of dopaminergic (DA) neurons in the substantia nigra is the neuropathological hallmark of the disease. The majority of PD cases are sporadic; however, familial forms have also been reported. Over the past decade, mutations linked to familial forms of PD have been identified in a number of genes such as alpha-synuclein (also known as SNCA) (Polymeropoulos et al., 1997), leucine-rich repeat kinase 2 (LRRK2) (Paisán-Ruíz et al., 2004), parkin (also known as PARK2) (Kitada et al., 1998), PTEN-induced putative kinase 1 (PINK1) (Valente et al., 2004), DJ-1 (also known as PARK7) (Bonifati et al., 2003), and ATP13A2 (Ramirez et al., 2006). Among these, alpha-synuclein and LRRK2 mediate autosomal dominant forms of PD, and the others mediate autosomal recessive forms. Discovery of these PD-linked genes has enabled an understanding of the molecular mechanisms underlying familial PD pathology, providing valuable insight into the pathological mechanisms involved in sporadic cases.
Mitochondrial dysfunction has been heavily implicated in PD pathogenesis (Henchcliffe & Beal, 2008). The activity of complex I, a major component of the mitochondrial respiratory chain, is decreased in substantia nigra and other tissues in PD patients (Keeny et al., 2006; Parker et al., 2008). Moreover, several complex I inhibitors successfully reproduce key features of PD such as loss of DA neurons and motor deficits. Exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) causes parkinsonism in humans (Langston et al., 1983). Administration of rotenone or paraquat also induces selective loss of DA neurons and produces locomotor defects in various animal models (Betarbet et al., 2000; Coulom & Birman, 2004; Cicchetti et al., 2005). Recent findings that parkin and PINK1 have critical roles in maintaining mitochondrial function and integrity have suggested that mitochondrial dysfunction is the prominent cause of PD pathogenesis, enabling investigation of the pathological mechanisms of PD at the molecular level (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). The following is a brief review of the recent findings related to the roles of parkin and PINK1 in mitochondria.
Parkin is critical in maintaining mitochondrial integrity
Parkin is an E3 ubiquitin ligase encoded by parkin, the most commonly affected PD gene conserved in various organisms from Drosophila to humans (Shimura et al., 2000). Parkin is composed of an ubiquitin-like domain in its N-terminus and two RING-finger domains in its C-terminus. In mammalian cell-based studies, Parkin can ubiqutinate and degrade several proteins including CDCrel-1 (Zhang et al., 2000), parkin-associated endothelin receptor-like receptor (Pael-R) (Imai et al., 2001), and cyclin E (Staropoli et al., 2003). From these results, endoplasmic reticulum (ER) stress resulting from accumulated Parkin substrates was proposed as the cause of DA neuronal death by loss of parkin. However, further studies failed to establish a meaningful relationship between these putative Parkin substrates and PD pathogenesis. To overcome the limitations of the cell-based study, several groups generated and characterized parkin null animal models. Although parkin null mice could not reproduce human PD symptoms, Drosophila parkin mutants demonstrated obvious phenotypes including locomotive defects and DA neuron degeneration (Greene et al., 2003; Pesah et al., 2004; Cha et al., 2005). In addition, administration of L-DOPA substantially rectified the behavioral defects of the parkin mutants, further confirming that parkin mutant fly models successfully parallel human PD patients (Cha et al., 2005). These mutants also showed defective wing posture and a crushed thorax. Histological examination of the parkin mutants demonstrated indirect flight muscle degeneration, which probably contributed to the locomotive defects along with DA neuron degeneration. Furthermore, mitochondrial swelling was found in the indirect flight muscles of the parkin mutants, suggesting that mitochondrial dysfunction may be an important cause of PD. However, these data cannot confirm whether mitochondrial swelling is a primary or secondary effect of parkin mutation. Further evidence is needed to confirm the importance of mitochondrial dysfunction in PD pathogenesis.
PINK1 and Parkin act in a common pathway in mitochondrial protection
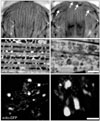
PINK1 is a serine/threonine kinase localized to the mitochondrial membrane via a mitochondrial targeting motif in its N-terminus (Valente et al., 2004). Most of the currently reported mutations are located in its kinase domain, indicating that PINK1 kinase activity is required for its role in PD protection (Klein & Lohmann-Hedrich, 2007). Interestingly, PINK1 fly mutants demonstrated phenotypes remarkably similar to parkin mutants, including flight disability, slow climbing speed, indirect flight muscle degeneration and a reduced number of DA neurons (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). Moreover, mitochondrial swelling was also observed in the indirect flight muscles and DA neurons (Fig. 1). Upon further genetic analysis, over-expression of mitochondrial protein Bcl-2 was found to rescue mitochondrial dysfunction and defective phenotypes in PINK1 mutants, indicating that mitochondrial defects are the main cause of PD-related phenotypes in PINK1 mutants (Park et al., 2006).
Because of the marked phenotypic similarities between parkin and PINK1 mutants, subsequent Drosophila genetic analysis were performed to test whether PINK1 and Parkin act in a common pathway. Transgenic expression of parkin markedly ameliorated the phenotypes of PINK1 mutants; however, parkin mutant phenotypes could not be recovered by over-expression of PINK1 (Clark et al., 2006; Park et al., 2006; Yang et al., 2006; Fig. 2). These data established that PINK1 and Parkin are linked in the same pathway to protect mitochondrial integrity and function with Parkin acting downstream of PINK1. In addition to the Drosophila results, over-expression of parkin successfully rescued the mitochondrial dysfunction induced by PINK1 knockdown in the mammalian system, demonstrating that the PINK1-Parkin pathway is conserved in flies and mammals (Exner et al., 2007).
Following the establishment of the PINK1-Parkin pathway, researchers have been trying to investigate the relationship between these two proteins. Using human DA cells and Drosophila models, Kim et al. demonstrated that PINK1 translocates Parkin to mitochondria in its kinase activity-dependent manner (Kim et al., 2008). Further analysis suggested that PINK1 phosphorylates Parkin on its linker region and promotes its mitochondrial translocation. Additional studies showed that PINK1 selectively translocates Parkin to impaired mitochondria, confirming the PINK1-dependent Parkin translocation that Kim et al. reported (Matsuda et al., 2010; Narendra et al., 2010).
PINK1 and Parkin remodel mitochondria
Since fly genetic analysis clearly demonstrated that the PINK1-Parkin pathway is involved in the protection of mitochondrial integrity and function, various efforts have concentrated on investigating the particular role of this pathway in mitochondria. Recent fly genetic studies showed that the PINK1-Parkin pathway regulates the mitochondrial remodeling process including mitochondrial fusion and fission. Mitochondria are dynamic organelles that constantly fuse and divide. Uncontrolled fusion-fission processes lead to severe damage of mitochondria morphology and function, causing the impairment of various cellular process and even cell death (Chan 2006). Surprisingly, in Drosophila, over-expression of Drp1, a guanosine triphosphatase (GTPase) for mitochondrial fission, or down-regulation of Opa1 or Marf expression, GTPases for mitochondrial fusion, rescued PINK1 and parkin mutant phenotypes, suggesting that the PINK1-Parkin pathway regulates mitochondria remodeling by promoting mitochondrial fission (Deng et al., 2008; Poole et al., 2008; Yang et al., 2008; Park et al., 2009). Therefore, loss of PINK1 or parkin may induce uncontrolled mitochondrial remodeling leading to severe mitochondrial dysfunction and subsequent DA neuronal degeneration. Results from other human cell-based studies, however, are not in agreement. Co-expression of PINK1 and Parkin, or expression of mitochondria-targeted Parkin induces mitochondrial aggregation in human DA neuroblastoma cells (Kim et al., 2008). Moreover, mitochondrial fragmentation was found in human neuronal cells lacking PINK1 or in the primary cells from human patients with PINK1 mutations (Exner et al., 2007; Kim et al., 2008; Wood-Kaczmar et al., 2008). To account for these different patterns in mitochondrial remodeling, researchers are attempting to dissect the exact mechanism involved in PINK1 and Parkin-mediated mitochondrial remodeling.
In addition to balancing between mitochondrial fusion and fission processes, the PINK1-Parkin pathway may also be involved in another mitochondrial remodeling process: mitophagy, the specific autophagic turnover of mitochondria. Narendra et al. reported that Parkin is selectively translocated to damaged mitochondria upon treatment of mitochondria damaging agents, and induces the turnover of damaged mitochondria through an autophagy-related gene 5 (ATG5)-dependent mechanism (Narendra et al., 2008). This finding elicited further studies to test whether PINK1 is required for the mitophagy-promoting activity of Parkin. In human DA neuroblastoma cells, co-expression of PINK1 and Parkin induces the formation of perinuclear mitochondrial clusters surrounded by autophasic vacuoles (Vives-Bauza et al., 2010). Moreover, knock-down or knock-out of PINK1 successfully inhibits mitochondrial damage-induced mitophagy (Matsuda et al., 2010; Narendra et al., 2010). Further analysis determined that PINK1 is selectively stabilized on impaired mitochondria, and activates the autophagic degradation of these mitochondria by recruiting Parkin (Matsuda et al., 2010; Narendra et al., 2010). These data provide the functional link between PINK1, Parkin, and the selective autophagy of mitochondria, suggesting that PINK1 and Parkin maintain mitochondrial integrity and function by selectively removing impaired mitochondria.
Mitochondrial targets of the PINK1-Parkin pathway in mitochondrial remodeling
In biochemical studies using Parkin proteins containing disease causing mutations, the ubiquitin ligase activity of Parkin is critical for normal physiological activities. Moreover, Parkin-mediated mitochondrial ubiquitination was observed in mitochondrial damaging agent-treated cells. Over-expression of dominant negative ubiquitin mutants prevented Parkin-induced mitophagy, demonstrating that ubiquitination links between Parkin and mitophagy (Geisler et al., 2010; Lee et al., 2010; Matsuda et al., 2010). To identify a putative Parkin target on mitochondria, molecular weight changes of various mitochondrial proteins upon mitochondrial damaging-agent treatment were evaluated. Surprisingly, a molecular weight shift of voltage-dependent anion channel 1 (VDAC1) was observed. Further biochemical analysis identified VDAC1 as a target for Parkin-mediated mitochondrial ubiquitination and subsequent mitophagy (Geisler et al., 2010). The ubiquitin-binding autophagic component p62 and HDAC6 were also essential for Parkin-dependent mitophagy (Geisler et al., 2010; Lee et al., 2010), suggesting that ubiquitinated VDAC1 may be an important adapter molecule for p62 and HDAC6. In addition, Ziviani et al. reported that the pro-mitochondrial fusion protein Marf is ubiquitinated by Parkin, inferring that Parkin prevents refusion of damaged mitochondria to support proper mitophagic degradation (Ziviani et al., 2010). These mitochondrial Parkin targets further confirmed the role of the PINK1-Parkin pathway in mitochondrial remodeling, suggesting a mitochondrial quality control system driven by PINK1 and Parkin (Whitworth & Pallanck, 2009). Under mitochondrial damaging stress, PINK1 selectively translocates Parkin to impaired mitochondria. In mitochondria, Parkin ubiquitinates its targets, prevents refusion of damaged mitochondria, and finally removes damaged mitochondria to protect mitochondrial function and integrity. These novel Parkin targets have only been studied in cell-based PD models and are yet to be confirmed in animal PD models. Therefore, further studies are required to confirm and refine the molecular mechanisms of PINK1 and Parkin with regards to regulating the mitochondrial remodeling process.
Concluding remarks
After the cloning of PD-associated genes, their roles in protection against PD were investigated in cell-based PD models. However, pioneering work using a genetic fly model revealed that the most affected PD gene, parkin, acts downstream of another PD gene, PINK1, to protect mitochondria, confirming the link between mitochondrial dysfunction and PD pathogenesis. Additional studies based on this finding revealed the exact role of the PINK1-Parkin pathway in mitochondria: balancing between mitochondria fusion and fission, and selective mitophagy of damaged mitochondria. Further studies investigating the molecular mechanisms of PINK1 and parkin in regulating mitochondrial remodeling will provide an enhanced understanding of PD pathogenesis and the development of an effective treatment strategy for PD.

 XML Download
XML Download