

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Multiple congenital anomalies-hypotonia-seizures syndrome-2 (MCAHS2) is an X-linked disorder comprising severe developmental delay, facial dysmorphism, neonatal hypotonia, early onset epileptic encephalopathy, neurologic disability and congenital abnormalities. PIGA germline mutation was shown to cause MCAHS2.1 The PIGA gene product is involved in the first biosynthesis of a protein called glycosylphosphatidylinositol (GPI) and remodelling of the GPI-protein complex, which plays a particularly important role in neurogenesis and embryogenesis.
Paroxysmal nocturnal hemoglobinuria (PNH) has been reported as a complement-medicated haemolytic disease caused by PIGA mutations on erythrocytes.2 Although somatic mutations in PIGA can lead to abnormalities in hematopoietic cells, germline mutations in PIGA have been reported to lead to an embryonically lethal disorder involving neurodegenerative encephalopathy and multiple congenital anomalies.3 Reccent advanced genome-wide screening approaches incorporating diagnostic exome sequencing have enabled genetic disorder to be detected in patients with rare and puzzling abnormalities and developmental delay.4
Data from seven patients with the germline PIGA mutation c.1234C>T (p.Arg412) have been published until now.15 Here we describe one neonate presenting with hypotonia, early onset epileptic encephalopathy and multiple congenital anomalies, who carried the c.1234C>T (p.Arg412) mutation. Although genetic testing was performed by chromosomal analysis and array comparative genomic hybridization CGH (aCGH), PIGA mutation was discovered by diagnostic exome sequencing. To our knowledge, this is the first report of a neonatal case with a germline PIGA mutation in Korea.
Case
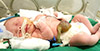
A 38-week, full term, appropriate for gestational age, male neonate with a 3,420 g birth weight was delivered by Caesarean section because of omphalocele noted on fetal ultrasonography. Prenatal ultrasound showed bilateral hydronephrosis and omphalocele. He required resuscitation and intubation after birth. His Apgar scores were 2 at 1 minute and 4 at 5 minutes. His birth weight was 3,420 g (50-75 centile), length was 49.5 cm (75-90th centile), and head circumference was 36 cm (>90th centile). On physical evaluation, he had omphalocele; a dysmorphic facial appearance; a depressed nasal bridge; a high-arched palate; upslanted palpebral fissures; a funnel chest; and hypospadias (Fig. 1). He also showed multiple joint contractures with both knee, ankle, elbow and wrist. He underwent surgery for correction of his reddish omphalocele (5 cm by 5 cm) at day 2 after birth. He was initially hypotonic, and developed myoclonic seizures at day 14 after birth. His electroencephalography (EEG) showed burst suppression pattern. Brain MRI showed a normal appearance. There was no evidence of any metabolic disorder. Serology tests for congenital infections were negative, and blood and cerebrospinal fluid cultures drawn at this time remained negative. We performed echocardiography to find out associated heart anomaly at day 0 and 2 mo of age, but there was no heart anomaly. His laboratory findings were as follows: white blood cell count 11,000/mm3, hemoglobin 13.8 g/dL and platelet count 369,000/mm3. His blood gases, electrolytes, blood ammonia, lactate/pyruvate, ketone, serum creatine kinase and glucose levels were within normal reference values. Abdominal ultrasonography showed left mild hydronephrosis. Sequence analysis of SMN did not identify any mutations related with spinal muscular atrophy. He did not show an amplification of a specific CTG repeat suggestive of myotonic dystrophy. The mother and his sister presented no clinical symptom. At 3 months, he resumed severe hypotonia, apnea and respiratory difficulties. Continuous positive airway pressure was initiated. His seizures were recurrent and resistant to multiple antiepileptic drugs. We used clonazepam, pyridoxin, valporate, and zonisamide for controlling seizure, but these were not effective. His conventional chromosomal analysis was normal (46, XY) and subsequent aCGH analysis showed normal results. However, the patient's dysmorphic face, which included congenital anomalies, hypotonia, and intractable seizures, suggested genetic disorders. Additional workup of exome sequencing analysis revealed a hemizygous nonsense mutation in PIGA (c.1234C>T, p.Arg412*). The patient is currently undergoing hospitalization for treatment of respiratory failure and recurrent seizures.
Patient's mother has one brother, who has two non-affected healthy child. We could not do exome sequencing analysis with patient's mother because of her refusal.
1. Exome sequencing analysis
Library preparation was performed with a TruSight One sequencing panel (Illumina, USA). This panel enriches for approximately 4,800 genes of clinical relevance. Massive parallel sequencing was conducted with a NextSeq instrument (Illumina). Local realignment and recalibration were performed using the Genome Analysis Tool Kit (GATK version 3.30). Among the 8,304 called variants, 1,375 remained after filtering for common variants with a minor allele frequency (1%) using multiple population databases (1000 Genomes Project, Exome Variant Server, and Exome Aggregation Consortium). Considering the phenotype of the patient, we concluded that a hemizygous nonsense variant (NM_002 641.3:c.1234C>T, p.Arg412*) in PIGA, a gene that has been associated with multiple congenital anomalies-hypotonia-seizures syndrome 2 (OMIM #300868), was the causative mutation. The variant was confirmed by Sanger sequencing (Fig. 2). The variant has previously been reported as pathogenic.
Discussion
MCAHS2 is X-linked disorder comprising neonatal hypotonia, dysmorphism, neonatal seizures, and multiple congenital anomalies with early infantile lethality. Here, we described the involvement of a pathogenic nonsense mutation in PIGA in MCAHS2.
The full name of PIGA is phosphatidylinositol glycan anchor biosynthesis class A. The product of this gene encodes a protein for the biosynthesis of the GPI anchors in the attachment of proteins to the plasma membrane during normal development.67 More than 150 proteins and 13 different genes have heterogeneous functions in a multistep process that produce GPI-protein complex.7 Specifically, phosphatidylinositol glycan class A has been identified as an important cause of a variety of multiple congenital anomalies and early-onset epileptic encephalopathies. Therefore, somatic mutations in PIGA can lead to the development of paroxysmal nocturnal hemoglobinuria in blood-forming cells, while germline mutations in PIGA gene can lead to the development of MCAHS2, Ferro-Cerebro-Cutaneous Syndrome (FCCS) and a less-severe form of encephalopathy.8910 We identified a hemizygous germline mutation (NM_0026 41.3:c.1234C>T, p.Arg412*) in the patient, in which, the 412th amino acid in PIGA (arginine) was replaced by a stop codon. A 1234C-T mutation in the last exon of PIGA resulted in an arg412-to-ter substitution.
The clinical characteristics of our case were very similar to those reported previously by Kato et al.11 The phenotype of our case could be classified into severe PIGA deficiency including refractory epileptiform discharges with a burst-suppression pattern on EEG, dysmorphic features and anomalies of other organs.12 Our case showed no anemia or paroxysmal nocturnal hemoglobinuria that is caused by somatic PIGA mutations. In contrast to other cases with mutations in PIGA, our case exhibited congenital malformation with omphalocele. To our knowledge, no known syndromes or reports have included both omphalocele and early onset epileptic encephalopathy. Van der Crabben et al.13 reported mildly elevated alkaline phosphatase levels and accelerated linear growth in patients with germline nonsense mutation in PIGA. In our case, the highest alkaline phosphatase level was elevated as 539 U/L at 5 weeks after birth.
MCAHS2 is a rare and devastating disorder in neonate. Furthermore, exact diagnosis of MCAHS2 is difficult, hindering early detection and treatment, which can lead to early lethality and developmental disorders. Our case study emphasizes how exome sequencing can be useful not only for the diagnosis but also for consultation of the genetic basis in the family. Further insights in diagnostic approach for MCAHS2 seems to be a crucial factor associated with minimal brain injury. Since the mutant PIGA appears to have variable pathogenesis according to retaining some activity, controlling PIGA levels would be helpful to the patient in the clinical course. In our patient, initial cytogenetic analysis and subsequent aCGH analysis could not easily identify this puzzling disorder that is suspected to be genetic in origin. Thus, this case highlights the important role of exome sequencing technology because it is more comprehensive than conventional cytogenetic assays and can reveal suspected genetic aberrations.4
In conclusion, MCAHS2 should be considered in any infant who presents with respiratory distress, hypotonia, and seizures with burst suppression pattern, regardless of symptoms severity. The most significant points for diagnosis are clinical suspicion for neonatal genetic disorder and a stepwise-genetic approach including exome sequencing is of particular value to uncover genetic disorders.

 XML Download
XML Download