

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glutamate is a principal neurotransmitter in most excitatory synaptic transmission while the synaptic glutamate receptors play a central role in the development and maturation of synaptic networks in the developing brain.12 Failure to synchronize the presynaptic glutamate release and the postsynaptic glutamate receptor-induced depolarization results in the defective synaptic neurotransmission, eventually leading to apoptosis or necrosis.34 Neuronal cell death, following hypoxic-ischemic (HI) injury, is caused by the complex cascades of pathophysiological events.4 Excitotoxicity by the overactivation of glutamate receptors in the discrete brain areas has been implicated as a major factor of central neuronal death in various pathologic conditions including HI injury.45
There are three subfamilies of ionotropic glutamate receptors; N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-S-methylisoxazole-4-propionic acid (AMPA) and Kainate.6 Kainate receptors (KARs) are tetrameric oligomers assembled from members of five different subunit classes: GluK1 (GluR5), GluK2 (GluR6), GluK3 (GluR7), GluK4 (KA1), and GluK5 (KA2).6 The pharmacological and functional properties of KARs differ depending on subunit composition. The synaptic activation of postsynaptic KARs and the presence of presynaptic KARs provide important mechanisms for regulating the excitatory and inhibitory synaptic transmission.7 They also play an important role in the developmental maturation of neuronal networks.1
There are accumulating evidences that the overstimulation of NMDA or AMPA receptor trigger the cascade involving HI injury.48 Intervention with neuroprotective agents may partially prevent HI injury. Several experimental approaches to salvaging brain tissue from neuronal death have been widely investigated.910 It has been also suggested that KARs may mediate glutamate-excitotoxicity in subpopulation of neuron.11 However, the researches for KARs has been hindered due to the lower expression levels of KARs and the lack of selective agonists and antagonists for discriminating between the AMPA and kainate subtypes.12 There is a need for a selective KARs because many agonists and antagonist for non-NMDA receptors were also active at the AMPA receptor subunits.13 Steady efforts for elucidating KARs function and structure had sustained over the past three decades, including molecular cloning of subunits, recombinant receptors and isolation of soluble fragment from each subunits.2714 Development of a new generation of selective agents has further expanded the understanding of the physiological and pathological properties of KARs.14 Among those compounds, SYM 2081 [(2S, 4R)-4-methylglutamic acid], is highly selective for the KARs compared to the AMPA and the NMDA receptors.15 Compared to the AMPA and NMDA receptors, it displays almost 3,000- and 200-fold selectivity for KARs, respectively.15 Also, it appears to act as a high-potency KAR-selective ligand whose active mechanism desensitizes KARs and thus acts functionally as a KAR antagonist.16 Therefore, SYM 2081 seems to be relevant to a probe to examine the physiological functions of KARs and the prototype of a novel class of therapeutic agents.16
As yet, the expression patterns of KARs in the neonatal HI brain injury have not been clearly established. There is also little report concerning the neuroprotective effect of SYM 2081 in the neonatal HI brain injury. Therefore, this study was designed to investigate the expression patterns of KARs and to determine the potential of SYM 2081 as a neuronal rescue agent after HI brain injury.
METHODS
1. Animal Experiment Experiment
This study was performed in accordance with the approved animal use guidelines of the Catholic University of Daegu. A modification of Levine preparation was used as a model for perinatal hypoxic-ischemic brain injury as previously described.17 Seven-day-old Sprague-Dawley (SD) rat pups were prepared for surgery. The midline of the neck was incised at the longitudinal plane under ether. The left carotid artery (LCA) was permanently doubly ligated with 5-0 surgical silk. Total time of surgery never exceeded 5 min. Following a 1 hour period of recovery with the dam and feeding, the pups were placed in plastic chambers, which were submerged in a 37℃ water bath and exposed to hypoxia (mixture of 8% O2 and 92% N2) for 2 hours. After this hypoxic exposure, the pups were returned to their dams for the indicated time. SYM 2081, purchased from Tocris Cookson (Bristol, UK), was prepared in phosphate-buffered saline (PBS) and injected intraperitoneally with dose of 10 mg/kg at 30 minutes before hypoxic exposure.
The animals were divided into five groups. Normoxia (N, n=4) group was subjected to neither ligation nor hypoxia. Hypoxia (H, n=4) group was exposed to hypoxia without operation. Hypoxia with Sham-operation (HS, n=4) group was subjected only to open-close surgery without LCA ligation. Hypoxia with LCA ligation-operation (HO, n=6) group was subjected to hypoxia without SYM 2081 injection. HO treated with SYM 2081 (HM, n=6) group was injected with SYM 2081. Pups from each litter were randomly assigned and marked to an N, H, HS, HO, and HM groups. Pups were killed at 7 days after hypoxic exposure and then the brains were extracted. The percentages of hemispheric areas were measured using a densitometer (Multi Gauge Software, Fuji Photofilm). The area ratio of left to right hemispheres was calculated.
2. Embryonic cortical neuronal cell culture of rats
Cultures of dissociated cortical neuronal cells were prepared from SD rat embryos pregnant for 19 days using methods similar to those previously described.18 The fetal pups were washed in 100% ethanol and Hanks' balanced salt solution (HBSS) (GibcoBRL, Grand Island, NY, USA). Cerebral cortices were removed and dissected at 37℃ HBSS containing 1 mM sodium pyruvate and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4), briefly. The dissected brain cortical tissues were then placed in 2 mL trypsin and incubated at 37℃ water bath for 1 min. After washing with 10 mL HBSS, the cell suspension was centrifuged at 1,000 rpm at 25℃ for 5 min and pellets were washed with HBSS (without phenol red). Cells were plated at approximately 2×106 cells/mm2 in each dish. Cells were cultured in Neurobasal media (GibcoBRL, Grand Island, NY, USA) (100 mL neurobasal, 2% B27 supplement, 0.25 mL glutamax I, 0.1 mL 25 mM glutamate, 0.1 mL 25 mM 2-mercaptoethanol) in a CO2 chamber. A fifth of the medium was replaced with fresh feeding Neurobasal media (GibcoBRL, Grand Island, NY, USA) (100 mL neurobasal, 2% B27 supplement, 0.25 mL glutamax I) every 3 days. The cortical neuronal cells were observed using inverted microscope (TS 100-F, Nikon instruments Inc., NY, USA) under high magnification (×200).
The cultured cells were divided into three groups: NC, normoxia; HC, hypoxia; HMC, hypoxia treated with SYM2081 (10 µg/mL). The NC group was prepared in 5% CO2 incubators while the other groups in 1% O2 incubators (94% N2, 5% CO2) for 16 hours.
3. RNA extraction
Brain samples were rapidly removed from the left cerebral hemispheres. Tissues and cultured cells were homogenized in TRIzol reagent (Invitrogen Corporation, Carlsbad, CA, USA) before a 5 min incubation at room temperature and chloroform treatment (200 µL) for dissociation of nucleoprotein complexes. The tubes were shaken by hand for 15 seconds and they were incubated at room temperature for 2 min. The upper aqueous (RNA) phase was collected following centrifugation (12,00x g for 15 min at 4℃). The aqueous phase was then collected into a fresh tube then precipitated into a gel-like pellet with 500 µL isopropyl alcohol by incubating for 10 min at room temperature and centrifuging 12,000x g for 15 min at 4℃. After removal of the supernatant, the RNA pellet was washed once with 1 ml 75% ethanol by centrifugation at 7,500x g for 5 min at 4℃. The RNA pellet was dried by air for 10 min before redissolving the RNA in 100 µL RNase-free water and incubating for 10 min at 60℃. The amount and purity of extracted RNA was quantitated by GeneQuant 1,300 spectrophotometer (GeneQuant™proRNA/DNAcalculator, GE Healthcare, Milwaukee, WI, USA). The RNA was then stored at -70℃ before further processing. For real-time polymerase chain reaction (PCR, for reverse transcription), total RNA (1 µg) was reverse transcribed for 1 hour at 37℃ in a reaction mixture containing 20 U RNase inhibitor (Promega, Madison, WI, USA), 1 mM dNTP (Promega, Madison, WI, USA), 0.5 ng Oligo (dT) 15 primer (Promega, Madison, WI, USA), 1x RT buffer and 200 U M-MLV reverse transcriptase (Promega, Madison, WI, USA). The reaction mixture was then incubated at 95℃ for 5 minutes to stop the reaction. The cDNA was then stored at -20℃ before further processing.
4. Real-time PCR for KARs
Real-time PCR was carried out in a 20 µL volume with 10 µL of kit-supplied iQ™ SYBR Green Supermix (Bio-rad Laboratories, Hercules, CA, USA), 1 µL (2 pmol) of each primer, 7 µL RNase-free water, 1 µL cDNA. Amplification was performed in 48 well PCR plates (Mini OpticonTM Real-Time PCR System, Bio-rad Laboratories, Hercules, CA, USA) with initial denaturation for 5 min at 95℃, followed by 40 cycles of denaturation for 30 s at 95℃, annealing 30 s at temperature indicated in Table 1, and extension for 30 s at 72℃. The final extension was for 10 min at 72℃. Real-time PCR data were analyzed with LightCycler software (Bio-rad Laboratories, Hercules, CA, USA). The experiment was repeated six times in the real-time PCRs.
5. Statistics analysis
The data were analyzed using the SPSS version 22.0 statistical analysis package. Examined data were assessed using the Student's t-test, general linear model, and analysis of variance. In each test, the data were expressed as the mean±SD, and P<0.05 was accepted as statistically significant.
RESULTS
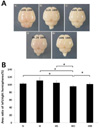
1. Gross changes in the neonatal HI brain injury (in vivo)
The area of the left hemisphere expressed as a percentage of that of the right hemisphere (Fig. 1). The area of left hemisphere showed similar to that of the right hemisphere in the N, H, and HS group. There was a reduction in the area of left hemisphere underwent LCA ligation compared to the contralateral, unligated side in HO group. On the other hand, under the same condition, administration of SYM 2081 before a hypoxic insult result in similar area percentages between both hemispheres in the HM group.
2. Microscopic changes in the embryonic cortical neuronal cell culture of rat (in vitro)
The cortical neuronal cells cultured in the normoxic environment (Fig. 2A) appeared normal, whereas those exposed in the hypoxic environment (Fig. 2B) appeared distinct morphological changes such as cell rounding and detachment from the growth surface. The affected cells treated with SYM 2081 in the HMc group (Fig. 2C) appeared similar to those in the Nc group.
3. The expressions of KARs in the neonatal HI brain injury (in vivo)
The expressions of GluK1, GluK3, GluK4 and GluK5 were increased in the H group compared to those in the N group whereas the expression of GluK2 was not increased in the H group (Fig. 3). The expressions of GluK1, GluK3, GluK4 and GluK5 were decreased in the HO group compared to those in the H group. These decreased expressions of GluK3, GluK4 and GluK5 were inhibited in the HM group. Interestingly, the expression of GluK1 was not increased in the HM group, but rather decreased compared to that in the HO group (Fig. 3A). Unlike other KARs, the expression of GluK2 was decreased in the H group compared to that in the N group and increased in the HO group compared to that in the H group (Fig. 3B). In the HM group, the expression of GluK2 was increased compared to that in the HO group.
4. The expressions of KARs in the embryonic cortical neuronal cell culture (in vitro)
The expressions of all KARs except for GluK2 were decreased in the Hc group when compared to those in the Nc group, whereas increased in the HMc group compared to those in the Hc group (P<0.05) (Fig. 4). The expression of GluK2 was not decreased in the Hc group compared to that in the Nc group (Fig. 4B).
DISCUSSION
In this in vivo study, gross morphological changes after the HI brain injury and the effect of SYM 2081 were demonstrated. In the N, H and HS group, the both side hemispheres were exposed to the same condition and therefore the area percentages between both hemispheres were similar. On the other hand, in the HO group, the left hemisphere was exposed to ischemic injury leading to permanent damage unlike right side.19 The area percentages of affected hemisphere were decreased as compared to those of the unaffected, contralateral hemisphere. Nonetheless, the affected hemisphere was restored comparable to size of the contralateral hemisphere by SYM 2081 treatment before a hypoxic insult. SYM 2081 decreased the parenchymal loss, observed in the affected hemisphere of the HO group. This result indicates that SYM 2081 exerts a potent protective effect against the HI brain injury.
KARs can induce rapidly desensitizing currents in response of agonists.20 While SYM 2081 function as agonist to open ion channel at certain concentration, it could function as antagonist to block these agonist-induced currents under continuous exposure at much lower concentration.16 It has been reported that parenteral administration of 200 mg/kg of SYM 2081 lead to ~6 µM steady-state brain concentration.16 Thus, the 10 mg/kg dose of SYM 2081 in this study would be expected to have much lower steady-state brain concentration. Therefore, SYM 2081 could function as antagonist for KARs at this dose.
In addition, this in vitro study showed similar effect such as the in vivo study. A substantial fraction of neurons underwent neuronal death in the Hc group compared to that in the Nc group and SYM 2081 treatment induced less neuronal death.
The expression of each KAR before and after a hypoxic insult also has been observed. As yet, there are no report of the expressions of the KARs' subunits after administration of SYM 2081 in the neonatal HI brain injury. Similar to NMDA or AMPA receptors, most KARs were activated after a hypoxic insult. It has been reported that the expressions of hippocampal GluK2, GluK3, and GluK4 were increased after adrenalectomy but reversed by corticosteroid or aldosterone treatment.21
The expressions of all KARs except for GluK2 were decreased in the HO group compared to those in the H group. In the in vitro study, the expression of all KARs except for GluK2 were decreased in the the Hc group compared to those the Nc group. Decreased expressions of several KARs in the HO group and the Hc group seems to be caused by the cell necrosis. Due to the significant cell loss, it is estimated that the expression patterns of KARs in the Hc group in in vitro study matches those in the HO group in the in vivo study. In the HM and HMc groups, these suppressions of GluK3, GluK4 and GluK5 were inhibited. This result indicated that increased expressions of KARs in response to hypoxia were sustained because SYM 2081 induced less neuronal loss.
Unlike other KARs, the expression of GluK2 was decreased in the H group compared to that in the N group and sustained in the HO and Hc group compared to that in the H and Nc group, respectively. In the HM and HMc groups, the expression of GluK2 was also increased compared to that in the HO and Hc group. This increase may also be a secondary response to less neuronal loss rather than a direct effect of SYM 2081. The expression of GluK2 seems to sustain even after the significant cell injury.
Interestingly, the expression of GluK1 in the ischemic hemisphere of the HM group was more suppressed than that of the HO group, indicating that SYM 2081 down-regulated expression of this subunit of KARs. Even in the in vitro study, the expression of GluK1 in cortical neuronal cells in HMc group was lower than that in the Nc group although increased in the Hc group. The expressions of GluK4 and GluK5 in the HMc group were lower than those in the Nc group although increased expressions than in the Hc group, but in the in vivo study, there were an increased expressions in the HM group compared to that in the N and HO group. In the in vivo study, only expression of GluK1 in the HM group was suppressed compared to that in the HO group.
As, SYM 2081 was known to its target as GluK1 and GluK2,1620 the expressions of GluK3, GluK4 and GluK5 may not be affected by SYM 2081. Although GluK1 and GluK2 are target for SYM 2081, in the HM and HMc group, the expression of GluK1 was suppressed and that of GluK2 was not suppressed and rather increased compared to that in the HO and the Hc group. So, the differential expression patterns between GluK1 and GluK2 in this study may not be elucidated.
GluK1 are mostly expressed in hippocampal and cortical interneurons as well as cerebellar Purkinje cells.2223 It has been reported that presynaptic KARs containing GluK1 regulate synaptic inhibition by GABA release.24 The mechanism of presynaptic inhibition occurs via a G protein-coupled cellular cascade.423 Depressed inhibitory synaptic transmission is associated with epileptogenesis and neuronal cell injury.25 Therefore, the suppressed GluK1 activation by SYM 2081 may lead to neuroprotection. One of the other possible explanation is that GluK1 has higher affinity and selectivity for SYM 2081 than GluK2 and GluK3.26 GluK1 and GluK2 was also implicated in the kainate-induced epilepsy model.27 In this model, the expression of GluK1 was significantly increased by kainate injection whereas no change in the expression of GluK2. Furthermore, the expression of GluK2 was increased after antiepileptic ketogenic diet.25 Rather the increased expression of GluK2 in the HM and HMc group would not signify the negative effect on neuronal survival. In other words, although SYM 2081 has target toward both GluK1 and GluK2, this study showed that this compound affect differently on expression of both KAR subunits. The degree of expression alone could not mean the functional activation. In case of GluK2, the expression was sustained after a hypoxic insult in both in vivo and in vitro study. So, SYM 2081 might modulate functional aspect rather than expression. On the contrary, SYM 2081 seems to suppress the enhanced expression of GluK1 in the HI brain injury.
It has been also reported that there are differences in response of agonists/antagonists between homomeric and heteromeric composition.28 Because GluK4 and GluK5 do not form functional homomeric channels, the composition of subunits of KARs shows different pharmacological properties to SYM 2081.1429 GluK1/GluK5 heteromers showed higher affinity to SYM 2081 than GluK2/GluK5 heteromers.29 The KAR expression and several modulatory mechanism such as Q/R editing have been shown to be developmentally regulated.7 GluK1 expression during development undergo the qualitative changes with peak expression around the period birth.22 GluK2 shows a prenatal expression peak in the cingulate gyrus of the neocortex.22 In this study, the expressions of KARs among the groups in P7 SD rat and E19 neuronal cell were compared. So, the developmental difference in the expression and modulation of each KAR together with duration and severity of a hypoxic insult may account for the differential expression patterns of some KARs as well as the discrepancies between in the in vivo and the in vitro study. The difference in injury type, duration of exposure and concentrations of SYM 2018 affecting cells may affect this discrepancies in expressions of each KARs between in the in vivo and the in vitro study.
This study may suggest that the degree of downregulation of KARs expression correlate with a necrosis from the HI brain injury and SYM 2081 treatment lead to less neuronal loss after a hypoxic injury with preservation of some KARs activation which was not antagonized by SYM 2081. KARs have distinct properties unlike other glutamate receptors in that they has ion-dependent gating30 and can signal through G protein like a metabotropic receptor.12 In addition, they can change their functional property by forming various tetrameric combinations of subunits, undergoing editing, alternative splicing, and transient interaction with binding proteins.17 Therefore, the limitation of this study is that the expression of each KARs alone cannot explain the subunit composition, distribution, and functional aspect of KARs. Nevertheless, the modulation of KARs expression treated with SYM 2081 in this study means that at least this compound might exert some neuroprotective effect in the HI brain injury.
In conclusion, this study demonstrated that SYM 2081 down-regulated expression of GluK1 and had little effect on the expression of GluK2. Additionally, SYM 2081 could prevent the degeneration of neonatal cerebral neuronal cells caused by a hypoxic insult. Accordingly, SYM 2081 could be neuroprotective effects on the HI brain injury in the neonatal rats through modulating KARs expression. The present study may prove useful for the further development of clinical strategies for perinatal HI encephalopathy induced by asphyxia.




 XML Download
XML Download