

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chordoid glioma (CG) is an uncommon, slow-growing primary brain neoplasm, first described as a distinct clinicopathologic entity by Brat et al. (1) in 1998. CG was named for its histologic features, which resemble those of chordomas expressing glial fibrillary acidic protein (GFAP) (1). It was incorporated into the World Health Organization classification as a grade 2 lesion under the category of glial tumor of uncertain origin. CG exclusively involves areas of the anterior portion of the third ventricle near the hypothalamus (2). The tumor is termed as "CG of the third ventricle" because of this stereotypical location, and only a few cases of extraventricular CG have been reported (34). To the best of our knowledge, 23 cases of suprasellar CG have been reported to date, including the current case. Of these, only a few cases of CG were localized to the suprasellar region without ventricular involvement (4). Although a few previous reports have described the typical radiologic and histopathologic features of CG, none of the reports focused on the radio-pathological correlation. This is the first report providing a comprehensive radio-pathological correlation of a pituitary macroadenoma-like extraventricular CG unusually located in the sellar and suprasellar regions.
CASE REPORT
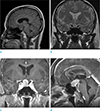
A 57-year-old woman presented with a 2-month history of headaches and visual disturbance. Neurological examination revealed bitemporal hemianopsia. Head computed tomography (CT) demonstrated a well-circumscribed oval mass without calcification or cystic components in the sellar and suprasellar regions. It appeared hyperdense and showed strong homogenous enhancement (Fig. 1a, b).
Magnetic resonance imaging (MRI) revealed a 2.8 × 1.5 × 1.7 cm mass in the pituitary fossa and suprasellar region, which was elevating and compressing the optic chiasm and the floor of the third ventricle superiorly without tumor extension. The normal pituitary gland and stalk were not discernible. The tumor was hypointense on T1-weighted images, iso- to hyperintense on T2-weighted images, and hyperintense after gadolinium administration (Fig. 2a-d). Presumptive diagnoses were pituitary macroadenoma, meningioma, or craniopharyngioma.
The patient underwent surgery via the transcallosal approach. During surgery, the mass appeared to originate from the diaphragma sellae behind the optic chiasm, and it was not abutting the third ventricle. Moreover, the pituitary stalk was encased by the tumor. The mass was partially resected because of the difficult surgical approach to the posterior portion of the mass. Histologically, the tumor was composed of epithelioid-to-spindle cells arranged in clusters and cords, embedded in a myxoid matrix (Fig. 3a). The neoplastic cells showed strong diffuse immunoreactivity for GFAP (Fig. 3b), CD34, and vimentin. A small subset of neoplastic cells stained positive for S100, cytokeratin, and epithelial membrane antigen; however, staining for synaptophysin was negative. The Ki-67 index was approximately 15%. The differential diagnoses based on tumor histology were CG, chordoid meningioma, and chordoma. Meningioma and chordoma were excluded based on the strong GFAP expression; CD34 immunoreactivity also supported the diagnosis of CG. The Ki-67 index reflected a relatively high proliferation rate, compared to previously reported rates.
Partial improvement in visual function was noted post-operatively; however, the patient developed memory and gait disturbance. Postoperative CT revealed a residual mass and hydrocephalus. The patient underwent ventriculoperitoneal shunt placement and received adjuvant gamma knife radiosurgery. The hydrocephalus stabilized, but the last available follow-up MRI scan obtained 5 months after the initial resection revealed disease progression with tumor growth.
DISCUSSION
First, Brat et al. (1) identified a tumor of the third ventricle presenting with both glial and chordoid features in 1998, and a new rare clinicopathological entity was named as "chordoid glioma of the third ventricle" (3).
Since then, to our knowledge, approximately 84 cases have been reported. The tumor develops mainly in adults, with a 1.7:1 female predominance, and mean age at presentation is 44.9 years (5). The most common symptom is headache; other symptoms include visual disturbance, memory disturbance, and endocrine dysfunction.
CGs exhibit remarkably similar characteristics on brain imaging (2); the tumor exclusively involves areas of the anterior portion of the third ventricle near the hypothalamus. A few exceptional cases with unusual locations such as the juxtaventricular region, left temporoparietal cortex, and thalamic pulvinar area (3) have been reported. On CT, the lesions are well circumscribed and hyperattenuating, with uniform contrast enhancement. On MRI, CGs are isointense on T1-weighted images, with avid and uniform contrast enhancement after gadolinium administration, and iso- to hyperintense on T2-weighted images. This significant contrast enhancement of CG is thought to arise from a blood-brain barrier breakdown and the ensuing extravasation of contrast medium, rather than tumor angiogenesis, which is confirmed by the lack of vascular proliferation on histological examination (6). On sagittal images, infundibulum shows posterior displacement by the tumor and greatest diameter of this tumor is usually observed in the superoinferior orientation (2). Intralesional calcification is rare. Cystic changes are occasionally observed (2). CGs are typically well-circumscribed and non-infiltrative tumors that are confined to the ventricle, but a rare case of tumor invasion into the optic chiasm has been reported (7).
Typical histopathologic findings of CG include the following: 1) clusters and cords of oval-to-polygonal epithelioid tumor cells with abundant eosinophilic cytoplasm, embedded in a PAS-positive mucinous matrix; 2) prominent lymphoplasmacytic infiltrates and Russell bodies throughout the tumor; 3) mitotic activity and anaplastic features (nuclear polymorphism, endothelial proliferation, necrosis) are absent or rare; and 4) minor tendency to infiltrate the surrounding brain parenchyma; however, reactive astrocytes, Rosenthal fibers, and chronic inflammatory cells are present in adjacent brain tissue (1).
Although many published cases have shown consistent histopathologic and radiological features of CG, no report has specifically discussed the correlation between typical imaging features and the histological features of CG. The CGs are usually hyperdense on non-enhanced CT images. We believe that the hyperdensity is probably related to the increased cellularity of these lesions, owing to prominent lymphoplasmacytic infiltrates. Moreover, the relative iso-to hyperintensity of CGs on T2-weighted images may reflect the presence of abundant eosinophilic cytoplasm in tumor cells and the abundant mucinous matrix.
The precise pathogenesis of CGs remains unclear. However, several recent ultrastructural studies have proposed an ependymal origin for these tumors (358). In the 3 cases studied by Cenacchi et al. (8), the authors suggest that CGs may be an ependymoma subtype, based on the ultrastructural similarities between CGs and the specialized ependymal cells of the subcommissural organ, which is prominent during embryologic life but undergoes a progressive involution. Pasquier et al. (5) proposed the term chordoid ependymoma of the lamina terminalis area, based on the ultrastructural findings and remarkably consistent anatomic location of this tumor in the anterior part of the third ventricle, closely associated with the lamina terminalis, near several circumventricular organs.
Of note, in the current case, neuroimaging studies revealed that the tumor appeared as a solid sellar and suprasellar mass, independent of the third ventricle, and appeared to mimic pituitary macroadenomas. Moreover, the epicenter of the mass as appeared to originate in the sella, and not from the lamina terminalis in the anterior third ventricle. To the best of our knowledge, 23 cases of suprasellar CG have been reported. Of these, only a few cases of CG were localized to the suprasellar region without ventricular involvement (4), as seen in this case. Furthermore, only one case was reported to be localized to the sellar and suprasellar regions without ventricular involvement, mimicking pituitary macroadenoma, as observed in our case (4). On MRI, classic CG can be clearly distinguished from the pituitary gland and stalk, excluding the possibility of pituitary adenoma (6). However, in the current case, the tumor could not be distinguished from the pituitary gland and stalk, prompting the preoperative diagnosis of pituitary macroadenoma. This unique location of current tumor leads to confusion with other sella and suprasella pathologies.
Various pathologic entities can arise in the sellar or suprasellar region, and common etiologies of masses involving both the sellar and suprasellar spaces include pituitary macroadenoma, craniopharyngioma, Rathke cleft cyst, and meningioma (9). Pituitary adenomas are the most common lesion involving the suprasellar region. Although their characteristics of signal intensity or enhancement are not distinctive, features such as sellar enlargement and erosion, cavernous sinus invasion, and lobulated margins are helpful in diagnosing pituitary adenomas (9). Craniopharyngiomas often have degenerative calcification and a prominent cystic component. In addition, In addition, one-third of craniopharyngiomas are heterogeneously hyperintense on T1-weighted MRI scans and show heterogeneous contrast enhancement in the solid portions and cyst wall. In contrast, CGs rarely show calcification, and stereotypically show homogenous contrast enhancement (9). On CT, CG may be confused with meningioma owing to its hyperdensity, and homogenous strong enhancement after contrast administration. Moreover, the relative isointensity of CG is similar to that of meningioma in T2-weighted images. However, the bilaterally symmetric posterior extension of vasogenic edema into the optic tracts may facilitate the differential diagnosis (2). Moreover, meningiomas frequently create an obtuse angle with the tuberculum or dorsum sellae (67%), show the dural tail sign or meningeal enhancement (68%), and are commonly associated with hyperostosis when the sella is involved (34%) (9).
CGs are reported to have poor prognosis. It was reported that 74% of patients with CG died from postoperative complications or tumor recurrence; the most common cause of death was pulmonary embolism (10).
Subtotal resection is associated with the risk of recurrence. Adjuvant radiation therapy has been performed in a limited number of cases; Kobayashi et al. (10) reported effective residual tumor control with gamma knife radiosurgery in three patients.
We present an unusual case of CG located in the pituitary fossa and suprasellar region without involvement of the third ventricle, which appeared to mimic pituitary macroadenoma. We recommend that the differential diagnoses for lesions in the sellar and suprasellar regions should be extended to include CGs in the list of possible etiologies.


 XML Download
XML Download