

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Pride and prejudice
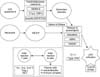
Lupus nephritis (LN) is a representative clinical feature of systemic lupus erythematous (SLE), which is scientifically challenging to comprehend its nature. The pathogenesis of LN involves a variety of pathogenic mechanisms. The pathogenesis of LN implicates altered cell death including aberrant apoptosis and formation of neutrophil extracellular traps (NETs) in breaking tolerance, the significance of autoantibodies, the role of the complement cascade, the contributions of adaptive immunity cross-talked with the innate immune system, genetic associations and various environmental factors in driving renal damage (Figure 1).
In spite of a knotted skein slowly being unraveled in return for endless efforts of researchers in various fields, we still haven't figured out the exact cause, which prevents us from achieving the goal of cure. In addition, some results have not been repeated in following studies and not been validated yet. For example, the intrarenal etiology of LN includes the several paradigms contemporarily in conflict with each other such as anti-double strand DNA (dsDNA) antibodies that cross-react with inherent renal antigens, anti-dsDNA antibodies targeting exposed chromatin in glomeruli, and relative antibody avidity for dsDNA, chromatin fragments, or cross-reacting antigens. In addition, LN patients have been reported to have increased numbers of apoptotic glomerular cells compared to healthy controls, in correlation with anti-dsDNA antibody levels, complement consumption, and cell proliferation while there are also evidences about a decrease in apoptotic cells from the glomerulus and tubulo-interstitium in LN biopsies compared to control kidneys. Therefore, I'd like to suggest that you stop here if you are eager to find the right answer to the pathogenesis of LN through this review. And for those who are only interested in LN rather than the whole story of SLE, the pathogenesis specific to LN is written in Italics.
Epidemiology-‘The nuclear bomb targets the kidney in SLE’
SLE is a systemic autoimmune disease with multi-organ inflammation by production of pathogenic autoantibodies directed against nucleic acids and their binding proteins and immune complexes reflecting a global loss of tolerance [1]. The prevalence of SLE ranges from 1.4% to 21.9%; incidence is estimated to be 7.4~159.4 cases per 100,000 person-years [2]. In Korea, the prevalence was reported to be around 20 per 100,000 populations and there are approximately 12,000 patients under treatment [3]. Most patients are female and younger than 50 years of age. However, male patients have a high incidence of nephropathy and greater severity of disease [4]. With the advent of advanced therapies, the 5-year survival rate has shown continuous improvement from 50% in 1953~1969 to nearly 90% to date [56].
LN is present in approximately 60% of SLE patients, with 25%~50% of patients presenting with clinical renal disease at the time of diagnosis [7], when occurring early in the course of SLE, is considered a major predictor of poor prognosis [8]. Patients with LN also have a higher standardized mortality ratio (6~6.8 vs. 2.4) and die earlier than SLE patients without LN [9101112].
MAIN SUBJECTS
How wolves attack human
1) Aberrant cell death and dead cell handling-‘Climbing to ALPS’
(1) Apoptosis
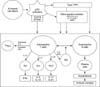
Accelerated cell death in SLE can potentially overwhelm the host clearance mechanism, resulting in the accumulation of apoptotic debris. And these changes contribute to induction of autoantibodies and other aberrant immune responses in SLE and in LN specifically [13]. Secondary necrotic cells release nuclear autoantigens that can lead to immune complex formation (Figure 2).
Some apoptotic signaling molecules have been reported to be related to SLE. One of these was the identification of mutations in Fas receptor and Fas ligand in mice [141516] and in humans that develop autoimmune lymphoproliferative syndrome (ALPS) [1718].
Defects in phagocytosis have been observed in SLE [19]. SLE patients have an accumulation of apoptotic cells in lymph node germinal centers likely due to the reduction in tangible body macrophages that specialize in the removal of dead cells [19]. Defects in the differentiation of myeloid progenitors into macrophages may potentially lead to phagocytosis defects in SLE [20].
Apoptosis-induced post-translational histone modifications are targets for autoimmune system in SLE [212223]. Microparticles from SLE patients which contain apoptosis-related histone modifications activate plasmacytoid dendritic cells (pDCs) and myeloid DCs (mDCs) that results in the induction of proinflammatory cytokines such as type I interferon (IFN) [24].
Some apoptotic signaling molecules including B cell lymphoma 2 (Bcl-2), Bim, transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), B cell-activating factor (BAFF), phosphatase and tensin homolog (PTEN), and p53 have also been linked to LN [25].
Whether increased cell death of glomerular cells is an important source of circulating and/or tissue nucleosomes promoting glomerulonephritis is controversial [26]. LN patients have been reported to have increased numbers of apoptotic glomerular cells compared to healthy controls, in correlation with anti-dsDNA antibody levels, complement consumption, and cell proliferation [27]. But, there are also evidences about a decrease in apoptotic cells from the glomerulus and tubulo-interstitium in LN biopsies compared to control kidneys [28]. In addition, renal cells from LN patients had enhanced proliferation without an increase in apoptosis. It is not clear why there are significant inconsistencies in determining if renal cells from LN patients undergo increased apoptosis, but one potential explanation could be the type of experimental method used to quantify this process [2930]. On the other hand, the deposition of glomerular ubiquitinated histone H2A was reported in a significant proportion of LN [3132].
(2) NETs
Enhanced formation and defective clearance of NETs contributes to SLE, especially renal disease (Figure 2). Neutrophils can extrude a meshwork of nuclear material bound to neutrophil granular proteins which mediates cleaving histones and promoting chromatin decondensation [33]. NET induction and clearance may result in a protective antimicrobial effect but excessive NET formation and inefficient removal could lead to tissue damage and autoantigen modification and externalization [34]. SLE patients are more prone to form NETs than neutrophils from healthy controls [35363738]. And SLE patients have an impaired ability to degrade NETs and proposed that this impairment contributes to the development of LN [39404142]. NET derived self-DNA complexed with neutrophil-derived antimicrobial peptides activatepDC Toll-like receptor (TLR) 9 and induce IFN α [38].
The renal biopsy analysis from patients with LN revealed the presence of NETs and infiltrating netting neutrophils in the glomeruli [35], which positively correlates with higher levels of circulating autoantibodies and enhanced activity index in kidney biopsies. Deoxyribonuclease (DNase) I is the major endonuclease found in circulation involved in degrading NETs. The correlation between DNase I deficiency and increased prevalence of LN was confirmed in SLE patients with renal involvement [394143].
2) The troops betray me
(1) Innate immunity
TLR: Persistent apoptotic debris containing nucleic acids can stimulate the inflammatory response through the activation of nucleic acid recognition receptors such as members of the TLR [44]. In SLE, TLRs might become aberrantly activated in the absence of foreign molecules [45] and associated with pDC activation and type I IFN production (Figure 3) [3638]. Indeed, TLR7 (receptor for single strand RNA) and TLR9 (receptor for DNA) mRNA expression was upregulated in PBMCs from SLE patients and levels correlate with the expression of IFN α [4647]. TLR7 was preferentially increased in SLE patients with antibodies against RNA-associated antigens, while TLR9 induction correlated with anti-dsDNA antibody titers [48]. And the upregulation of TLR7 was observed when healthy neutrophils were cultured with sera from SLE patients with active disease [36]. SLE patients with active disease had a higher number of TLR9 expressing B cells and monocytes than did patients with low disease activity, and levels of these cells correlated with levels of antibodies to dsDNA [49]. In TLR9 deficient lupus-prone mice, the generation of anti-dsDNA and anti- chromatin autoantibodies was specifically inhibited [50].
The nucleic acid component of immune complexes also activates intrarenal inflammation by TLRs in intrarenal macrophages and DCs to produce large amounts of proinflammatory cytokines and IFN [5152535455565758]. In pristane-treated mice, TLR7 was specifically required for the production of RNA-reactive autoantibodies and for the development of glomerulonephritis [59]. Studies of pharmacologic or genetic manipulation of TLR7 expression or function support a central role for TLR7 in inflammation, loss of tolerance, and type I IFN production [60616263]. And the activation of TLR3 on antigen presenting cells (APCs) or renal mesangial cells can aggravate LN by recruiting polymorphonuclear cells to the site of inflammation, where they can contribute to renal injury [6465]. Genetic variants of TLR3 (receptor for dsRNA), TLR7/8 and TLR9 have been associated with LN. These variants may contribute to severe renal insufficiency in LN. In addition, signaling through particular TLR9 genetic variants was associated to more severe renal disease at the time of LN presentation [6667].
Cytokines: Levels of many cytokines are elevated in SLE such as IFN, TNF, interleukin (IL)-4, IL-6, and IL-10 and their main effects are the promotion of autoantibody production and inflammation (Figure 3).
Type I and II IFNs have emerged as key cytokines in the pathogenesis of SLE and increases in their levels precede autoantibody development [68]. Upregulation of TNF can increase type I IFN expression [6970]. IFN α, a type I IFN has multiple effects consistent with known immunologic features of SLE, such as upregulation of BAFF, decreased regulatory T (Treg) cell function, and induction of plasma cells. In particular, the prevalence of type I IFN signaling was higher in T cells than in other immune cell types in patients with SLE [71]. A direct pathogenic role for IFN in mouse models of lupus is also supported by studies in which exogenous administration of IFN α exacerbates disease [7273].
Patients with SLE may also have an imbalanced T cell cytokine profile characterized by decreased IL-2 [74] and increased IL-17 levels [75]. Production of IL-2 is impaired on multiple levels [74]. IL-2, in addition to being critical for Treg cell development and function, is also necessary for restricting expression of IL-17. In SLE, IL-17 may mediate local tissue damage through the induction of inflammatory cytokines and chemokines, and by recruiting other immune cells. The differentiation of the T helper cell subset producing IL-17 is dependent on IL-23, and an anti-IL-23 antibody ameliorated disease in one mouse model of lupus [76].
B cell activation and autoantibody production are promoted in SLE by BAFF. Serum levels of BAFF are increased in patients with SLE and positively correlate with autoantibody titers [77]. Transgenic overexpression of BAFF in a mouse model of lupus exacerbated disease [78], BAFF is a critical factor for B cell homeostasis and high BAFF levels might reduce the stringency of B cell selection, allowing autoreactive clones to persist in the periphery [79].
Following immune complex deposition, a large variety of inflammatory mediators is produced in LN kidneys with spreading of the response as disease progresses [8081]. A Type I IFN signature is also a feature of LN kidneys [8283]. IFN has multiple detrimental effects on the kidneys including vascular rarefaction and injury to glomerular parietal cells and podocytes [8485]. Examples include CCL2, a chemokine expressed early in the glomerulonephritis process, and TNF that is expressed at proteinuria onset [8687]. Multiple cytokines such as IFN γ, IL-21 and IL-17 have also been detected in LN kidneys [88]. Once tissue injury occurs, soluble products released from injured cells amplify the inflammatory response by stimulating extracellular and intracellular innate immune receptors [8990919293]. Nevertheless, not all renal inflammatory mediators are necessary for the inflammatory process. For example, IL-17 deficiency alters the course of LN only in some models in which Th17 cells infiltrate the kidneys [9495]. IL-4 drives signal transducer and activator of transcription 4 (STAT4) activation, which leads to autoantibody production. Autoantibody-mediated pathology in LN is supported further by genetic variants within the T follicular helper (Tfh) differentiation pathway [9697], where IL-6 and IFN γ [98] via STAT [99] activate TfH differentiation.
Complements: The complement systems affect the ability of innate immune cells to facilitate phagocytosis of nuclear antigens and cell debris. One of the most remarkable genetic associations in SLE is the early components of the complement system classical pathway [100101102103]. More than 90% of patients with homozygous deficiency of C1q are reported to have SLE and the high titers of autoantibodies are observed in more than 70% of these patients [100101102104]. About 10%~30% of homozygous C2-deficient patients develop SLE [100105106]. Arthritis, malar rash, discoid rash, and photosensitivity are seen in the majority of C2-deficient patients with SLE [100, 101103104106]. Complete homozygous deficiency of C4 is rare but, more than 75% of these patients develop this disease. Approximately 50% of SLE-C4-deficient patients develop LN and more than 70% has antinuclear antibodies and anti-Ro autoantibodies in their serum [100101104].
Patients with C1 deficiencies usually present SLE at an early age, in similar female:male proportions, with severe symptoms and prominent cutaneous manifestations [107]. Anti-C1q antibodies which target a neo-epitope of bound C1q are present in 2%~8% of the healthy population, but they are present in 30%~48% of patients with SLE [108]. Their titer correlates to active renal disease with a sensitivity of 44%~100% and a specificity of 70%~92% [109].
The complement system is generally activated in LN and can directly mediate kidney injury through the terminal pathway, or indirectly increase renal inflammation by recruiting leukocytes to the kidney. In a lupus cohort, 23% of patients had autoantibodies to C1q and to C3b [110]. Anti-C3b and anti-C1q levels tended to increase in the months leading up to renal flare [111]. LN has been associated genetically with deficiencies in the opsonin C1q, C2 and C4 [112], Genetic variants of C-reactive protein (CRP) [113] and mannose binding lectin [114], also contribute to LN by disrupting complete clearance of autoantigens, enhancing inflammation, and increasing autoantibodies to C1q.
(2) Acquired immunity
B cells: The absolute number of B cells is not different to that of controls in SLE patients. But, certain peripheral B cell subsets in SLE patients showed differently compared to healthy controls by the following mechanisms (Figure 3).
Loss of tolerance and altered B cell differentiation in SLE might present from birth or acquired as part of the disease process [115]. Human studies have clearly implicated loss of B cell tolerance. Early immature B cells show increased levels of autoreactivity in SLE, possibly owing to a break in central B cell tolerance [116]. Patients with inactive SLE fail to remove B cells expressing self-reactive B cell receptors (BCRs) expressed by naïve B cells due to defects of selection against autoreactive B cells [117]. Activation of B cells through the TLR pathway or cytokines such as BAFF promotes loss of tolerance. Mouse models have demonstrated that transitional B cells are susceptible to accelerated maturation by TLR 9, which bypasses tolerance checkpoints [118119120121]. In addition, IL-10 secreting B cells with regulatory capabilities show functional impairment in SLE [122123].
High number of self-reactive mature naïve B cells which subsequently originate autoantibody producing plasma cells is the most reported characteristic of the abnormal B cell homeostasis in SLE characterized by the expansion of peripheral plasmablasts [124], which also correlates with disease activity and the titer of autoantibodies [125]. And the pool of memory B cells is enlarged. Since these cells have low activation thresholds, they present a risk for autoimmunity and the regulation by FcγRIIb receptors may be inhibited [126]. So these cells can be rapidly activated in a non-antigen-specific manner by the combination of TLR agonists and a proliferation-inducing ligand (APRIL) (TNFSF13A) or BAFF (TNFSF13B) as well as by the combination of cytokines, such as IL-21 and BAFF [77127].
Anti-dsDNA antibodies react with several renal cell types and are thought to be central to the nephritis process. The relative amount of anti-dsDNA antibodies has been calculated to comprise up to 20% of the total eluted immunoglobulin (Ig)G from nephritic kidney [128129130131132]. There are two theories about the pathogenic process about dsDNA antibodies. First, anti-dsDNA antibodies recognize exposed chromatin in the mesangium or in glomerular basement membrane. DNA specific B cells are stimulated by chromatin fragments and histone specific T helper cells. The emerging anti-dsDNA antibodies bind exposed chromatin in glomeruli and initiate LN [128133]. The completely lostof renal DNase I during progression of SLE seems to reduceclearance of chromatin from dead cells, and to promote harmful accumulation of undigested chromatin in glomeruli [133134135]. Others indicate that antibodies target cross-reacting antigens that appear as normal constituents in glomeruli [133136] or that chromatin- IgG complexes derive from circulation [137138139]. The B cells specific for chromatin or inherent glomerular structure such as laminin or entactin, respond by producing cross-reactive anti-dsDNA/anti-chromatin antibodies. These antibodies may bind exposed chromatin fragments or homologous, inherent antigens in kidneys, lungs, and other organs [133136]. Autoantibodies to annexin1 and α enolase have also been detected in LN kidneys [140]. Meanwhile, Infiltrating leukocytes form de novo lymphoid organs inside the kidney, which involve the clonal expansion of B cells. Such B cells undergo intrarenal proliferation and activation, which contributes to local inflammation and tissue pathology in addition to their role for systemic and intrarenal autoantibody production [141142]. B cells derived from human LN biopsies recognize vimentin, an intracellular structural protein that is cleaved and extruded from apoptotic cells [143]. Serum anti-vimentin antibodies are associated with decreasing GFR and increasing tubulointerstitial damage, and are associated with severe interstitial disease in LN [143144].
There are genetic variants that affect B-cells to break tolerance, secrete autoantibodies that contribute to kidney damage in LN. Genetic variants in the BCR complex and proximal signaling molecules are enriched in SLE patients and may contribute to LN [145]. The SLE patients with genetic variation of CSK has amplified inhibitory phosphorylation of Lyn, thus increasing BCR-mediated activation of mature B cells [146]. And these patients carries many lupus-associated autoantibodies that contribute to LN [147]. Genetic variants of CD40 which positively regulates B-lymphocyte activation through the adaptor molecule TRAF6 are associated with LN [148]. CD40 synergizes with TLRs and the BCR allowing to further drive immune dysregulation associated with SLE and renal disease in LN [149].
T cells: Loss of T cell tolerance through multiple mechanisms exists in SLE. There is aberrant signaling through the T cell receptor (TCR) in patients with SLE. In T cells from patients with SLE, the CD3ζ chain (which mediates signaling via tyrosine-protein kinase ZAP 70) is down regulated, causing ZAP 70 to be replaced by FcRγ. FcRγ then pairs with tyrosine-protein kinase SYK rather than with ZAP 70, resulting in hyperactivation of the TCR signaling pathway [150151]. Despite this hyperactivated phenotype, T cell production of IL 2 is actually impaired [74152].
Patients with SLE also show altered T cell subset populations (Figure 3). Th17 cells found infiltrating the kidneys of patients with lupus nephritis, and in the skin lesions of patients with SLE [153]. Double-negative T cells (CD4−CD8−) are expanded in patients with SLE [154155] and seem to be the primary source of IL 17 in SLE [156]. And these T cells are thought to contribute to loss of tolerance [154155], as they also express IL 1β and IFN γ, and promote B cell differentiation and antibody production.
Both B cells and T cells from LN kidneys are clonally expanded, and the same T cell expansions have been detected in the peripheral blood [143157] and in the urine of LN patients [158]. Aberrant T cell–B cell interactions are also observed in SLE [159160]. The pathologically expanded and activated TfH cell compartment markedly affects B cell differentiation. And expansion of the TfH cell subset correlates with increased disease activity and severity in patients with SLE [161162163]. The expansion of TfH cells in SLE may be directed by interaction with OX40 ligand (also known as TNF ligand superfamily member 4 [TNFSF4]), which is expressed on myeloid antigen-presenting cells [164]. Genetically determined increased OX40L expression promotes human SLE by effector T cells proliferation and plasma cell development. Loss of B cell OX40L ameliorates the SLE through declining in TfH cell numbers [165].
T cells from LN kidneys are clonally expanded, and the same T cell expansions have been detected in the peripheral blood [143], particularly IL-17 producing CD3+/CD4+ or CD3+CD4/8−/−T cells [156]. And multiple T cell cytokines such as IFN γ, IL-21 and IL-17 have also been detected in LN kidneys [88]. TfH cells can be seen within lymphoid aggregates in kidney biopsy samples from patients with active LN, and activated TfH cells correlate with autoantibody titers in these patients [96166].
Circulation-‘the vessels are tossed and turned’: Endothelial cells produce Platelet-derived growth factor (PDGF)-B whose interaction with PDGF-Rβ on mesangial cells is required for the development of glomerular disease. Expression of PDGF isoforms is upregulated in many forms of renal injury, causing mesangial hyperproliferation, matrix production, cytokine and chemokine release, and renal fibrosis [167]. Podocytes and endothelial cells also interact by bidirectional diffusion of cytokines/growth factors through the glomerular basement membrane [168]. And in diseased tissue, both activated glomerular endothelial cells and damaged podocytes release endothelin 1 that amplifies glomerular injury by causing mitochondrial stress [169].
And there are abnormal vascular function and tissue hypoxia in LN. The capacity for angiogenesis and capillary repair is lost owing to dissociation pericyte from capillaries and diminished production VEGF, leading to capillary infarction in both the glomerulus and the interstitium [170171]. Other disturbances of angiogenesis reported in LN include a decrease in the ratio of pro-angiogenic Ang1/anti-angiogenic Ang2, down regulation of the angiogenic factor FGF-2, an increase in the VEGF inhibitor ADAMTS-1, and alterations in endothelial nitric oxide synthase [172173174175]. Recent studies have shown that injured renal tubular cells have mitochondrial dysfunction, reprogram them to a pro-fibrotic phenotype, and contribute to their death [80]. Fibroblasts may contribute to tissue injury by producing pro-inflammatory mediators [176]. Fibrotic tissue may disrupt normal anatomic structures and interfere with oxygen diffusion, thus exacerbating hypoxia [177].
3) Genetics-‘ascribing everything to my parents?’
SLE is known to have a strong genetic link, with a heritability of 66%. Data suggest that concordance of SLE is 10 times more frequent in monozygotic than in dizygotic twins. The twin concordance rate for SLE is 25%~30% in monozygotic twins compared with 2% in dizygotic twins [178]. Most of genes associated with SLE are associated with multiple autoimmune diseases. The overall genetic risks identified to date are limited, with each gene generally conferring a relative risk <2 (Figure 4).
The rare but high-risk deficiencies in complement pathway gene products, including C2, C4, and C1q, are thought to contribute to lupus pathogenesis by impairing clearance of cellular debris [179180]. SLE develops in over 90% of C1q-deficient individuals [181]. Similarly, SLE development is strongly associated with C4 deficiency (75%) and to a lesser degree with homozygous C2 deficiency (10%~30%) [182]. However, the deficiency of these genes in patients with SLE is extremely rare.
Several genes have been associated with SLE susceptibility, most prominently in the human leukocyte antigen (HLA) loci [183184]. Then, FcγRIIA and FcγRIIIB which mediate the phagocytosis and immune function of the immune complex have been reported, In addition, CRP and integrin alpha M (ITGAM) are related [185, 186]. Mutation in the integrin α M (CD11b)-encoding ITGAM gene induced TLR-dependent proinflammatory signaling and IFNα signaling in lupus-prone MRL/Lpr mice [187].
HLA DRB1*1501 (DR2) and DR3 B1*0301 are class II alleles consistently shown to be associated with SLE [188]. More recently, a large genome wide association studies (GWAS) found that the best model for association was a combination of HLA alleles including B*08:01 and B*18:01 in class I, DQB1*02:01,DRB3*02:00, and DQA*01:02 in class II and a class III single nucleotide polymorphism (SNP) (rs74290525) located in SLC44A4 [184189]. Recent GWAS have superseded older candidate gene studies and have shown >40 genes associated with SLE outside of the MHC in European populations [189]. A large number of lupus-associated SNPs found in genes that encode proteins involved in induction of type I IFN and the innate immune response in SLE pathogenesis, such as IFN regulatory factor 5 (IRF5) and IRF7, TNF α-induced protein 3 (TNFAIP3) [190191192193]. And the expression of type I IFN signature genes such as Interferon α-inducible protein 27 (IFI27), interferon-induced protein 44-like (IFI44L), lymphocyte antigen 6 complex, locus E (LY6E), TNFAIP6, and interferon induced transmembrane protein 1 (IFITM1) in SLE patients was increased compared to other patients with autoimmune disease [194]. Additional lupus-associated variants that alter adaptive immune system activation are involved in cytokine signaling, such as STAT4, or efficiency of signaling downstream of the T and B cell surface antigen receptors, such as , protein tyrosine phosphatase, nonreceptor type 22 (PTPN22) in the case of both T and B cells, and LYN, B cell scaffold protein with ankyrin repeats 1 (BANK1), B lymphoid tyrosine kinase (BLK), TNFAIP3, and others in the case of B cells [195].
A GWAS in Koreans has been recently published [196]. Two loci were detected in 1174 SLE cases and the loci were, an intergenic SNP between FCH and double SH3 domains 2 (FCHSD2) and purinergic receptor P2Y2 (P2RY2), and autophagy related 16 like 2 (ATG16L2). A locus that was detected as suggestive, huntingtin interacting protein 1 (HIP1) is replicated in this study. None of these loci has been as yet detected in Europeans, but in Koreans, several European loci were confirmed once more. These were STAT1-STAT4, TNFSF4, TNFAIP3, IKAROS family zinc finger 1 (IKZF1), HIP1, IRF5, BLK, WDFY family member 4 (WDFY4), ETS proto-oncogene 1 (ETS1) and interleukin 1 receptor associated kinase 1 (IRAK1)-methyl-CpG binding protein 2 (MECP2) [196]. And GWAS was conducted in 4,478 SLE cohort from six East Asian conturies, general transcription factor II-I repeat domain-containing protein 1-general transcription factor 2-I (GTF2IRD1-GTF2I) being the most significant locus among ten new loci [197].
Recently, genetic risk factors have been identified as follows. NADPH oxidase-encoding Ncf1 gene SNP induced production of ROS, so increases the risk of developing SLE [198]. Mutation in the tartrate-resistant acid phosphatase (TRAP)-encoding ACP5 gene result in the expression of IFN-stimulated genes and the production of IL6 and TNF. And an excess of heterozygous ACP5 missense variants was observed in SLE [199]. Transcription factor Blimp1-encoding Prdm1 deficiency in DC led to modulated antigen presentation and the TfH cell repertoire to contribute to autoimmunity [200].
Carriers of HLA-DR4 and DR11 were protected against LN [183]. Conversely HLA-DR3 and DR15 conferred an increased risk of LN. In another approach, a meta-analysis of three GWAS was done to identify risk alleles for LN in patients already known to have SLE [201]. Here the most significant associations for LN mapped to the PDGF receptor A gene and the gene for the sodium-dependent glucose cotransporter solute carrier family 5 member 11 (SLC5A11). In LN, PDGF may mediate kidney cell proliferation, matrix accumulation, and intrarenal inflammation. Several SLC genes have been associated with chronic kidney disease (CKD) [202]. Variants in SLC5A11 may have a role in proximal tubule inositol reabsorption and mediate a decrease in serum and an increase in urine myoinositol [202]. Additionally, SLC5A11 may mediate apoptosis through the programmed cell death and TNF-pathways [203]. HLA loci were less strongly associated with LN in this analysis. Other risk genes include TNFAIP3 interacting protein 1 (ABIN1), TNFSF4, STAT4, ITGAM, kallikreins and FcγRIIIa low-binding alleles [185186204205206207208]. These results suggest a link between inflammation and LN as well as a contribution from pathways that regulate the renal response to inflammation and injury. However the relative risk associated with most of these genetic variants is low.
Epigenetic processes include DNA methylation, post-translational histone modifications were identified in SLE. T cells from patients with active SLE have global DNA hypomethylation [209], especially those from patients with LN [210]. And IFN-stimulated genes (ISGs) were specifically hypomethylated in patients with SLE [210]. Naive CD4+ T cells become primed for Th2, Th17, and TfH cell responses through the activity of the chromatin-modifying enzyme histone–lysine N methyl transferase EZH2 during a flare [211]. Post-translational histone modifications were shown to be aberrant in T cells from patients with SLE. These aberrations were corrected by treatment with mycophenolate mofetil [212]. And histone H4 acetylation was shown to be globally increased in monocytes from patients with SLE [213]. Changes in microRNA (miRNA) expression have been identified in peripheral blood mononuclear cells and renal tissue from patients with SLE [214215216]. MiRNAs identified in patients with SLE seem to affect pathways that affect TLR signaling and expression of ISGs [217218].
4) Environment-‘I hate my neighbor’
(1) Environmental factors-inside the body
Infection has been associated with the occurrence of SLE (Figure 4). Epstein–Barr virus (EBV) and cytomegalovirus are considered to be SLE triggers [219], whereas Helicobacter pylori [220], hepatitis B virus [221], and parasite infections are thought to be protective [222]. Viral infections induce IFNα release, which triggers antiviral immunity as well as lupus disease activity [223]. In addition, EBV activates B cells and contains amino acid sequences similar to those of Ro antigen, resulting in molecular mimicry [224].
The following additional data support the role of microorganisms in SLE. Lipopolysaccharide (LPS) is a component of the cell wall of Gram-negative bacteria that can activate TLR4. Serum levels of LPS are increased in patients with SLE [225] and biomarkers of LPS engagement by TLR4, such as shedding of CD14, correlate with disease activity [226]. Bacterial biofilms represent another mechanism by which microorganisms interact with the immune system. Many biofilms contain amyloid-DNA complex, so greatly increased the production of autoantibodies in lupus-prone mice [227]. Immune stimulation by bacterial DNA results from characteristic sequence motifs that center on an unmethylated cytosine and guanine (CpG) dinucleotide. Bacterial DNA which contain CpG motif more frequently than mammalian DNA presents a structural motif that represents a pathogen associated molecular pattern that can signal a pattern recognition receptor to trigger innate immunity via TLR9 [228]. The pDCs, which responds to TLR9, is a main source of type 1 IFN and therefore has been implicated in lupus pathogenesis [229230]. The microbiome which is the collection of bacteria, viruses, and fungi that coexist on and in the human body is related to SLE. In women with SLE, the ratio of Firmicutesto Bacteroidetes was seen lower than in healthy individuals, even during times of remission [231]. The mechanism of the effect is not fully understood, but certain gut bacteria foster the development of Treg cells [232233].
Furthermore, bacterial products stimulate intrarenal immune cells and renal cells, which can trigger a transient aggravation of proteinuria and kidney damage. Bacterial lipopeptide and LPS aggravate glomerulonephritis and potently induces severe albuminuria in MRL (lpr/lpr) mice [234].
(2) Environmental factors-outside the body
Ultraviolet (UV) light have long been recognized as contributors to SLE (Figure 4) [235]. UV drives apoptosis, providing an immunologic stimulus and increase in the load of dead cells by causing keratinocyte death [236]. And UV decreases DNA methylation level of CD4+ T cells in SLE patients [237238]. UV exposure can induce the secretion of IL-1 and TNFα in keratinocytes, mast cells and Langerhans cells, and can recruit and activate DCs, T cells and pDCs to release IFNα, further releasing chemotactic factors [239].
Drug induced SLE involves inhibition of methyl-transferases, a process that enhances the unmasking of endogenous nucleic acids and the activation of TLR7 and TLR9 [240241]. And UV light converts propranolol into a proinflammatory aryl hydrocarbon receptor ligand, possibly explaining its association with lupus-like disease [242].
Besides, silica exposure from a variety of industrial occupations and smoking is associated with an increased risk of SLE. A longer duration of exposure to silica dust is associated with greater risks [243]. And current smoking and >10 pack-years of smoking with anti-dsDNA positive SLE was observed [244]. And Vitamin D deficiency and zero minor allele of CYP24A1 significantly increased the risk of transitioning to SLE [245]. The alcohol consumption, on the other hand, decrease risk of SLE in large prospective Nurses' Health cohorts (Figure 4) [246].
5) Sex hormone-‘If I were born a man?’
SLE has a female preponderance of 10:1. The administration of estrogen to postmenopausal women doubles their risk of developing the disease and the inhibition of estradiol in patients with SLE with tamoxifen had modest beneficial effect on the disease [247]. Estrogen enhances immune responses through diverse mechanisms (Figure 1).
The prepubertal ovariectomy of female NZB x NZW F1 (B/W) mouse model for SLE was shown to reduce autoantibody levels, but had no effect on mortality. Conversely, orchiectomy of B/W male mice exhibited accelerated disease onset and shortened life span compared with intact male B/W mice [248]. Treatment with 17 β-estradiol accelerated disease in both female and male B/W mice, while disease was ameliorated in B/W mice treated with testosterone [249]. Treatment of R4A BALB/c mice with 17 β-estradiol resulted in a significant rise in anti-dsDNA antibodies, an increase in the number of anti DNA-secreting B cells [250]. and induced significant aberrations in the selection of autoreactive B cells into the mature B cell pool [251]. In addition, estrogen preferentially increased the proportion of marginal zone B cells [251], which participates in T independent immune responses. Estrogen treatment blocked BCR mediated apoptosis, which is essential for the elimination of autoreactive B cells [251252]. The antiapoptotic Bcl-2 protein, which is a known estrogen target [36], was increased in estrogen treated mice [250253]. An increase in Bcl-2 may enhance the survival of B cells that would normally be eliminated by tolerogenic signals [254].
In human SLE studies, the concentration of estradiol and prolactin in lupus patients was increased [255]. However, the level of these hormones is not related to the severity of SLE, and the concentration is within the physiological range. Females with SLE have even lower androgen levels than their normal female counterparts. The oxidation of androgens in SLE is increased when compared to males with the disease [256257]. The expression of intracellular ER-beta which is anti-inflammatory was reduced in T cells from SLE patients with SLEDAI-2K scores > 6 as compared to those with scores <6 or healthy controls. No difference was found for ER alpha differences [258].
Treatment of R4A BALB/c mice with 17 β-estradiol induced resulted rising in anti DNA antibody titers and immune complexes depositions in the kidneys [250]. And treatment of R4A BALB/c mice with tamoxifen prevented the estrogen-induced production of anti-DNA antibodies and immune complex deposition in the kidney [259].
CONCLUSION
Hope springs eternal
SLE is a systemic autoimmune disease with multi-organ inflammation. Accelerated cell death and formation of NETs activate pDC TLR 9 and induce IFN αcontributing to the development of SLE, especially LN. Many cytokines such as IFN, TNF, IL-4, IL-6, IL-10, and activated complement system enhanced the promotion of autoantibody production and inflammation. Loss of tolerance and altered B-cell and T cell differentiation in SLE might present from birth or acquired as part of the disease process of LN. Over 100 genes have been associated with susceptibility to SLE such as HLA, FcγRIIA and FcγRIIIB, ITGAM, deficiency of C2,C4, and C1q. Environmental factors such as infection (EBV, CMV, microbiome), UV light,smoking, some drugs, silica, Vitamin Ddeficiency increased the risk of transitioning to SLE. We are still far away from really knowing the pathogenesis of LN. However, the development of target therapies and an eventual cure will come in our lifetime.


 XML Download
XML Download