

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Inflammation is a normal physiological response to an infection or injury, such as aggression by microbes, trauma, or heat and radiation. Inflammation works to maintain homeostasis and is a highly regulated process with both pro- and anti-inflammatory components to ensure the prompt resolution of noxious conditions. In the initial stages of inflammation, macrophages destroy the abnormal stimuli, and remove the apoptotic bodies of the dead neutrophils as well as any remaining hazard factor. The macrophages then present the antigen to T lymphocytes to initiate the mechanisms of acquired immunity, which leads to the production of antibodies, cytokines and memory cells. The macrophage activity then switches from proinflammatory to antiinflammatory to remove any elements of aggression, thereby achieving homeostasis. Macrophages play a key role in the innate immune response and form a bridge between the innate and acquired immune response. In certain circumstances, however, when chronic inflammation is produced, macrophages may have a harmful effect and cause lesions. Therefore, inflammation is the classic “double-edged sword”, in which macrophages cut both ways. Activated macrophages have two different phenotypes related to different stimuli: M1 (classically activated) and M2 (alternatively activated). M1 macrophages are proinflammatory and play a key role in the host defense mechanism, while M2 are associated with the responses to anti-inflammatory reactions and tissue remodeling. The transformation of different phenotypes of macrophages regulates the initiation, development, and cessation of inflammatory diseases. An imbalance of macrophage M1∼ M2 polarization is often associated with a range of diseases or inflammatory conditions, such as rheumatoid arthritis and systemic lupus erythematous. (J Rheum Dis 2018;25:11-18)
Go to : 
REFERENCES
1. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005; 6:1191–7.

2. Kumar V, Cotran RS, Robbins SL. Robbins basic pathology. Philadelphia: Saunders;2003.
3. Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest. 2008; 118:413–20.
4. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008; 454:428–35.
5. Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macro phages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011; 51:267–88.
6. Hume DA. Differentiation and heterogeneity in the mononuclear phagocyte system. Mucosal Immunol. 2008; 1:432–41.
7. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005; 7:211–7.
8. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010; 32:593–604.
9. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005; 23:344–6.
10. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010; 11:889–96.
11. Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol. 2009; 175:2439–53.
12. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011; 12:231–8.
13. Narayana Y, Balaji KN. NOTCH1 upregulation and signaling involved in Mycobacterium bovis BCG-induced SOCS3 expression in macrophages. J Biol Chem. 2008; 283:12501–11.
14. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010; 70:4840–9.
15. Whyte CS, Bishop ET, Rückerl D, Gaspar-Pereira S, Barker RN, Allen JE, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol. 2011; 90:845–54.
16. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol. 2012; 189:3439–48.
17. Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999; 17:701–38.
18. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010; 106:1559–69.
19. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011; 121:2736–49.
20. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014; 513:559–63.
21. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a proangiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. 2014; 5:5350–68.
22. Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, et al. Re-po-larization of tumor-associated macrophages to proinflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012; 4:341–3.
23. Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. 2011; 286:1786–94.
24. Zhuang G, Meng C, Guo X, Cheruku PS, Shi L, Xu H, et al. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation. 2012; 125:2892–903.
25. Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006; 10:233.
26. Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage “switch” phenotype. Biomed Res Int. 2015; 2015; 341308.
27. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006; 116:1494–505.
28. Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol. 2008; 3:99–126.
29. Vandooren B, Noordenbos T, Ambarus C, Krausz S, Cantaert T, Yeremenko N, et al. Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum. 2009; 60:966–75.
30. Gerlag DM, Tak PP. Novel approaches for the treatment of rheumatoid arthritis: lessons from the evaluation of synovial biomarkers in clinical trials. Best Pract Res Clin Rheumatol. 2008; 22:311–23.
31. Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996; 39:115–24.
32. Tak PP, Bresnihan B. The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum. 2000; 43:2619–33.
33. Kennedy A, Fearon U, Veale DJ, Godson C. Macrophages in synovial inflammation. Front Immunol. 2011; 2011; 00052.
34. Hamilton JA, Tak PP. The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum. 2009; 60:1210–21.
35. Choi S, You S, Kim D, Choi SY, Kwon HM, Kim HS, et al. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J Clin Invest. 2017; 127:954–69.
36. Shin TH, Kim HS, Kang TW, Lee BC, Lee HY, Kim YJ, et al. Human umbilical cord blood-stem cells direct macrophage polarization and block inflammasome activation to alleviate rheumatoid arthritis. Cell Death Dis. 2016; 7:e2524.
37. Sestak AL, Fürnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann Rheum Dis. 2011; 70(Suppl 1):i37–43.
38. Kavai M, Szegedi G. Immune complex clearance by monocytes and macrophages in systemic lupus erythematosus. Autoimmun Rev. 2007; 6:497–502.
39. Orme J, Mohan C. Macrophage subpopulations in systemic lupus erythematosus. Discov Med. 2012; 13:151–8.
40. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006; 25:383–92.
Go to :
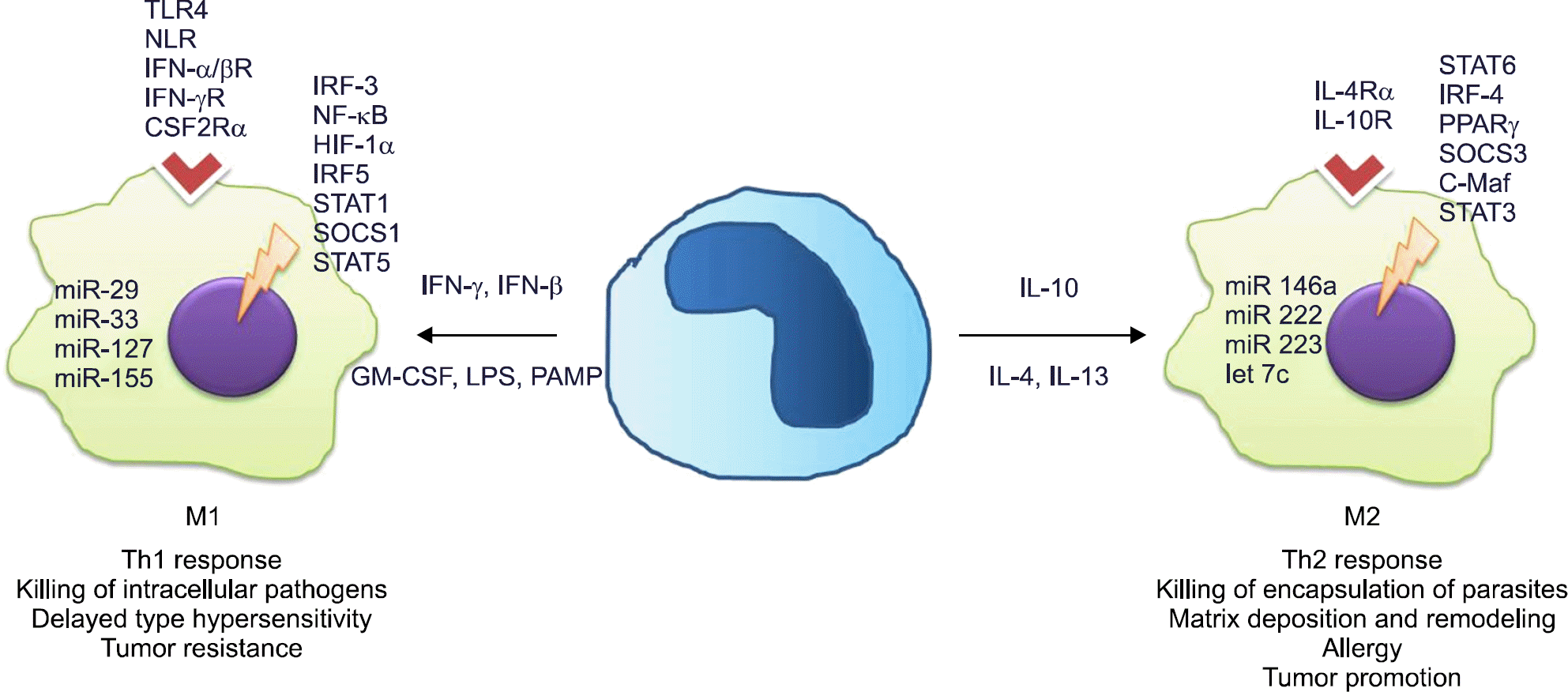 | Figure 1.M1 and M2 macrophage polarizing pathways. TLR: toll-like receptor, NLR: nod-like receptor, IFN: interferon, CSF2R: colony stimulating factor 2 receptor, IRF: interferon regulatory factor, NF: nuclear factor, HIF: hypoxia-inducible factors, STAT: signal transducer and activator of transcription, SOCS: suppressor of cytokine signaling, miR: microRNA, GM-CSF: granulocyte macrophage colony-stimulating factor, LPS: lipopolysaccharide, PAMP: pathogen-associated molecular patterns, IL: interleukin, PPAR γ: peroxisome proliferator activated receptor γ, C-Maf: cellular muscular aponeurotic fibrosarcoma. |
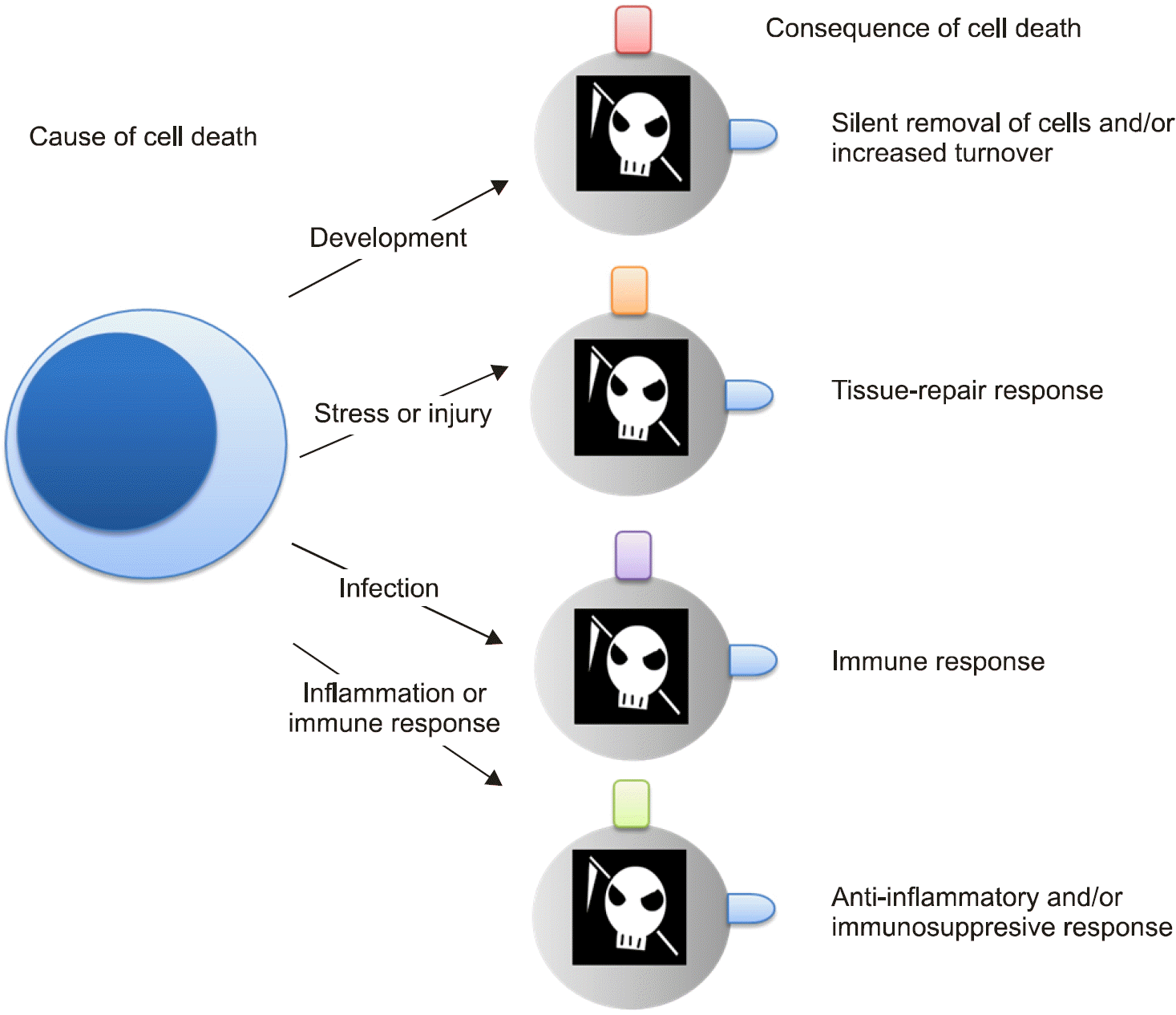 | Figure 2.Macrophage and apoptosis. Apoptotic cells are recognized by their lipid phospha-tidylserine (blue) on the plasma membrane and, subsequently, are removed by phagocytosis. In addition, apoptotic cells produce other signals (colored rectangles) that control the outcome of their recognition by macrophages, but its consequences are likely to rely on the cause of cell death. |
Table 1.
Subset of macrophage polarization
PAMPs: pathogen-associated molecular patterns, DAMPs: danger-associated molecular patterns, IFN: interferon, GM-CSF: granulocyte macrophage colony-stimulating factor, TNF: tumor necrosis factor, IL: interleukin, Arg-1: arginase-1, Chi313: chitinase3-like protein 3, TLR: toll-like receptor, TGF-β: transforming growth factor-β, VEGF: vascular endothelial growth factor.
 XML Download
XML Download