

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Rheumatoid arthritis is a chronic rheumatic disorder with a prevalence of 0.5% to 1% and is characterized by synovial inflammation and damage of joint and bone leading to deformity and disability [1]. Recently, biologic therapies targeting cytokines and inflammatory mediators have been developed for use in rheumatic diseases including rheumatoid arthritis, and have shown superior efficacy compared to pre-existing therapies; however, high costs, limited effectiveness in certain patient groups, and severe infectious complications limit their wide-spread use and reveal a need for the development of novel treatment [2].
In 1897, an association between the development of jaundice and amelioration of arthritic symptoms was first noted in juvenile rheumatoid arthritis patients [3], then in 1938, the study of Hench [4] noted a similar phenomenon in adult rheumatoid arthritis patients and showed that the effect occurred when the extent of jaundice was above a certain threshold and that the effect varied depending on the severity of jaundice [34]. Furthermore, the putative substance, which was hypothesized to be a chemical agent, was named “substance X” and components of bile such as bilirubin, bile salts, mucin, and lipids as well as abnormal hepatocellular substances resulting from tissue damage were initially suggested as candidate agents. Subsequently, Hench hypothesized that substance X was a corticosteroid and discovered its effectiveness as a treatment and thus was awarded a Nobel Prize [5]. However, further studies revealed that corticosteroids were not the chemical agents responsible for the effect of jaundice on arthritic symptoms and showed that the oral or parenteral administration of chenodeoxycholic acid (CDCA) ameliorated arthritic symptoms; thus, bile salts were suggested as the most likely candidate agent for “substance X” [67].
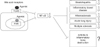
The cell surface bile acid receptor Takeda G-protein-coupled receptor 5 (TGR5) (also known as G protein-coupled bile acid receptor 1 [GPBAR1], membrane- type receptor for bile acids [M-BAR], and BG37) and the nuclear hormone receptors farnesoid X receptor (FXR; also known as NR1H4) were discovered and it was found that bile acids activate signaling pathways through these receptors to regulate bile synthesis, enterohepatic circulation, and triglyceride, cholesterol, energy and glucose homeostasis [8]. Recently, bile acids were found to regulate inflammation and mediate immune homeostasis [910111213141516]. Moreover, natural, semi-synthetic, and fully synthetic drugs that target these receptors were developed and are currently being studied for use in various inflammatory disorders such as alcoholic hepatitis, non-alcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis [1718]. On the other hand, experiments studying the effect of bile acid agonists on inflammation and immune regulation has been done mostly in in vitro or in vivo studies in mouse models and studies using human cell lines are relatively lacking and the diseases under study are mostly limited to inflammatory liver diseases, intestinal inflammatory diseases, atherosclerosis, acute lung injury, and multiple sclerosis, thus suggesting a need for further studies in rheumatic disorders (Figure 1).
The following review summarizes the expression profiles of bile acid receptors in various cells, tissues, and diseases, the roles of bile acid receptors in inflammation and immune response, and studies that look at the effects of bile acid receptor agonists in animal models of inflammatory disorders and in clinical trials, in order to argue for the therapeutic potential of bile acid agonists in inflammatory rheumatic disorders including rheumatoid arthritis.
MAIN SUBJECTS
Bile acids and its receptor
Bile acids are water-soluble amphipathic molecules that are synthesized in hepatocytes from cholesterol and secreted as the primary bile acids cholic acid (CA) and CDCA, which upon entering the intestinal lumen, are transformed by resident bacteria into the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA), then reabsorbed in the distal intestines and circulate back to the liver in a process known as enterohepatic recirculation [19]. Besides acting as detergents in lipid digestion and cholesterol absorption, bile acids also function through various intracellular signaling pathways via receptors such as FXR (NR1H4) or TGR5 (GPBAR1). Bile acids are conjugated with glycine or taurine before secretion from hepatocytes and these chemical features require transport proteins for bile acids to enter cells, and so intracellular bile acid signaling via the nuclear receptor FXR is specific to tissues expressing those transport proteins such as the small intestine and the liver [1819]. On the other hand, the cell surface receptor TGR5 does not require such transport proteins and may act on various types of cells and tissues [18]. The affinity for FXR is strongest for CDCA, then DCA, LCA, and CA, while for TGR5, LCA is the strongest, followed by DCA, CDCA, CA. Ursodeoxycholic acid (UDCA) and oleanolic acid are also TGR5 agonists [20].
Regulation of bile acid receptors expressions
FXR is expressed not only in tissues involved in enterohepatic circulation which include the liver and small intestine, but is also expressed in the kidneys, adrenal glands, lungs, and thyroids as well as monocytes and macrophages, osteoclasts; on the other hand, TGR5 is expressed in epithelial cells lining the gall bladder and bile duct, muscle, kidneys, brain, intestines, brown adipocytes, Kupffer cells, and monocytes [1721222324]. Research regarding the expression and regulation of TGR5 and FXR in monocytes and macrophages which play important roles in the pathogenesis of chronic inflammatory disorders is lacking; however, recent research found that in monocytes that were differentiating into dendritic cells, TGR5 expression was decreased, while in fully differentiated dendritic cells TGR5 expression was absent, and this decrease was related to granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin (IL)-4, which was in turn related to a decrease in TGR5 mRNA [22]. Compared to monocytes, macrophages which were induced to differentiate using M-CSF showed decreased TGR5 expression while those which were induced to differentiate using interferon (IFN)-γ did not show decreased TGR5 levels [22]. In one study, TGR5 binding by TGR5 agonists was shown to induce receptor internalization, but this result was unable to be replicated by other studies [2125]. While plasma membrane toll-like receptors (TLR 2, 4, 5, 6) were found to decrease FXR, intracellular TLRs (TLR 3, 7, 8, 9) were found to increase FXR, indicating that the regulatory effect on FXR expression is dependent on the type of TLR; TLR9 mediated FXR expression was found to be mediated by interferon regulatory factor (IRF) 7 [26]. In the liver, lipopolysaccharide (LPS)-induced inflammation was found to decrease FXR expression while in Hep-3B human hepatoma cells tumor necrosis factor (TNF)-α and IL-1β was found to decrease FXR expression [27]. Furthermore, in mouse and human macrophages, FXR was found to be downregulated by IFN-γ via signal transducer and activator of transcription (STAT)-1 dependent transcriptional repression, suggesting that various inflammatory mediators found in chronic inflammation can lead to a decrease in FXR expression and anti-inflammatory effects [28]. According to a preliminary study conducted by the authors of this review, synovial fluid macrophages isolated from RA patients expressed lower levels of FXR protein compared to peripheral blood monocytes from healthy subjects, which is consistent with the results of studies done in other chronic inflammatory disorders (unpublished data). However, studies looking at FXR and TGR5 expression and regulation in chronic inflammatory disorders including RA are lacking and further studies are necessary to better characterize their action.
Role of bile acid receptors in inflammation and immune response
1) TGR5
Ligand binding of TGR5 leads to the activation of adenylate cyclase and intracellular accumulation of cyclic adenosine 3′,5′-monophosphate (cAMP), leading to protein kinase A (PKA) activation and cAMP response element binding protein (CREB) phosphorylation [824]. TGR5 was found to be abundantly expressed in human and rabbit monocytes and macrophages, and bile acids were found to inhibit lipopolysaccharide-stimulated cytokine production in rabbit alveolar macrophages and THP-1 cells induced to express TGR5, suggesting TGR5 suppresses inflammation in monocytes and macrophages [25]. Furthermore, LPS-induced pro-inflammatory cytokine production was found to be suppressed by TGR5 stimulation in mouse macrophages and Kupffer cells, and was also found to be markedly increased in TGR5-/- mouse macrophages compared to wild type macrophages, and this effect was associated with a comparatively severe liver necrosis and inflammation, suggesting TGR5 mediates the anti-inflammatory effects of bile acids [1529]. The FXR/TGR5 dual agonist INT-767 suppresses histologic changes in nonalcoholic steatohepatitis and increases IL-10 as well as the proportion of anti-inflammatory intrahepatic monocytes [30]. Bile acids were found to induce the differentiation of monocytes to dendritic cells with an IL-12 hypo-producing phenotype, and this effect was induced by a TGR-5-specific agonist but not by FXR-specific stimulation, suggesting TGR5 induces the differentiation of monocytes to anti-inflammatory dendritic cells (DCs) [31]. In low density lipoprotein receptor (LDLR)-/- mice, TGR5 suppressed inflammation and decreased lipid accumulation, conferring protection to atheroma generation in the animal model of atherosclerosis [14]. Moreover, taurolithocholic acid (TLCA), a potent TGR5 agonist, induced nitric oxide (NO), which has anti-atherogenic effects, in sinusoidal endothelial cells (SEC) and human umbilical vein endothelial cells (HUVEC) and also suppressed TNF-α-induced adhesion of monocytes, vascular cell adhesion molecule-1 (VCAM-1) expression, and activation of nuclear factor-κB (NF-κB), and also suppressed LPS-induced monocyte adhesion to mesenteric venules in vivo, suggesting TGR5 suppresses inflammatory responses in endothelial cells [2932]. LCA, a potent natural TGR5 agonist, was found to be markedly decreased in patients with coronary atheroma, suggesting a decrease in LCA may contribute to the pathogenesis of atheroma in a TGR-5 dependent manner [33]. In a study using TGR5-/- mice as an inflammatory bowel disease (IBD) animal model, the extent of pathology was more severe in those mice compared to the wild type, and TGR5 agonist administration lead to a decrease in pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, IFN-γ in bowel tissue as well as a decrease in the extent of pathology [34]. Lamina propria mononuclear cells (LPMCs) isolated from the intestines of Crohn disease patients showed increased TGR5 expression levels, and decreased TNF-α expression was seen in CD14+ cells isolated from those patients when they were treated with TGR5 agonists, suggesting TGR5 is a regulator of inflammation in the bowel [22]. Furthermore, rs11554825, a TGR5 single- nucleotide polymorphism, is associated with primary sclerosing cholangitis and ulcerative colitis development, and homozygous individuals for the G allele of rs3731859 which is in perfect linkage disequilibrium (LD) with the risk allele C of rs11554825 demonstrate low TGR5 expression levels, suggesting TGR5 may be important in the development of primary sclerosing cholangitis and ulcerative colitis pathogenesis [35]. In the multiple sclerosis animal model, experimental autoimmune encephalomyelitis (EAE) mice, TGR5 agonist resulted in decreased activation of monocyte and microglial activation, decreased trafficking of monocytes and T cells into the central nervous system (CNS), and improvement of EAE clinical scores, suggesting TGR5 may be a potential therapeutic target in multiple sclerosis [16].
2) FXR
LPS-induced hepatic inflammation induced massive necrosis and inflammation in FXR-/- mice but not in wild type mice, and FXR agonists reduced hepatic inflammation and fibrosis in nonalcoholic steatohepatitis mice fed methionine and choline-deficient (MCD) diet, and FXR agonist treatment resulted in reduced IL-6 induced C-reactive protein expression in hepatoma cell lines [363738]. Furthermore, the use of a synthetic FXR agonist markedly decreased liver damage in a concanavalin A-induced autoimmune hepatitis animal model, and FXR-/- mice were more susceptible to concanavalin A-induced hepatitis than wild type mice [39]. The above findings suggest FXR has an important role in regulating hepatic inflammation. FXR-deficient mice exhibited more severe intestinal inflammation than wild type mice in studies that used colitis animal models such as dextran sodium sulfate (DSS)- and trinitrobenzenesulfonic acid (TNBS)-induced colitis models, and FXR stimulation resulted in a decrease in pro-inflammatory cytokines production and chemical-induced intestinal inflammation [910]. Moreover, LPS-induced TNF-α production was suppressed by INT-747 in human peripheral blood mononuclear cells, purified CD14 monocytes, dendritic cells, and LPMCs isolated from IBD patients [10]. In a study using the TNBS colitis animal model, TLR9 stimulation lead to decreased severity of colitis, the effect of which was found to be dependent on IRF7 binding and induction of the FXR promoter, suggesting FXR may mediate the housekeeping activities of TLR9 [26]. FXR is expressed in macrophages and vascular smooth muscle cells, and FXR stimulation has been shown to inhibit NF-κB activation and inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 expression, leading to reduced vascular inflammation and atherosclerosis, and FXR agonists resulted in reduced IL-1β, IL-6, and TNF-α expression in macrophages, leading to decreased atherosclerosis, suggesting FXR ligands may be effective in controlling inflammation in atherosclerosis [404142]. Another study showed INT-767 resulted in decreased pro-inflammatory cytokine production and circulating lipid levels in atherosclerosis [43]. In a study using the LPS-induced acute lung injury mouse model, FXR-/- mice showed increased susceptibility compared to control mice and increased inflammation and defective lung regeneration, while constitutive FXR activation lead to improvement of the lung injury repair defect, suggesting FXR has a protective role in inflammation-induced acute lung injury [44]. In a recent study involving the ovalbumin-induced acute murine asthma model, CDCA administration lead to reduced infiltration of cells into the airspace and peribronchial areas, goblet cell hyperplasia, mucus secretion and serum immunoglobulin E levels, as well as reduced T helper type 2 cytokines such as IL-4, IL-5 and IL-13, and TNF-α, suggesting FXR agonists may have therapeutic potential in allergic asthma [45]. In the EAE mouse model, FXR-/- mice exhibited increased disease severity compared to wild type mice, while FXR agonist administration resulted in reduced lymphocyte activation and cytokine production, suggesting FXR agonists may also have therapeutic potential in multiple sclerosis [12].
Regulatory mechanisms of inflammation and immune response by bile acid receptors
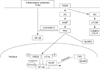
TGR5 activation leads to adenylate cyclase activation and cAMP-mediated cell signaling as well as changes in intracellular calcium concentration in certain cell types, which lead to intracellular signaling cascades [824]. Anti-inflammatory effects of TGR5 are mainly mediated by NF-κB signaling inhibition, which occurs via induction of β-arrestin 2 and inhibition of phosphorylation of inhibitory factor kappa B alpha (IκBα) or via c-Fos stimulation and phosphorylation, leading to c-Fos binding to NF-κB p65 and the formation of a c-Fos-p65 complex which is unable to bind the TNF promoter, suggesting TGR5 inhibits TNF-α via this mechanism [131522]. In another study using overexpression of the mutant of the mouse TGR protein (TGR5-A217P), which is unable to activate cAMP-CREB signaling, the cAMP-CREB signaling was found to be important in NF-κB inhibition and the inhibition of cytokine production [14]. Besides inhibition of NF-κB signaling, another study of macrophages found TGR5 inhibited LPS-induced inflammation via Akt stimulation and mammalian target of rapamycin (mTOR) complex 1 activation, which lead to an increase of the dominant-negative CCAAT/enhancer-binding protein β (C/EBPβ) isoform, liver-enriched inhibitor protein (LIP). Furthermore, bile acids were found to inhibit the Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome activation via the TGR5-cAMP-PKA axis, and lead to inhibition of LPS-induced sepsis and systemic inflammatory disease [1146].
Inhibition of NF-κB by FXR occurs by decreased DNA-binding by the transcription factor, similar to the mechanism of other nuclear receptors [36]. The nuclear receptor corepressor (NcoR) was found to be stabilized at the IL-1β promoter NF-κB responsive element of the IL-1β promoter by FXR activation, suggesting that NcoR stabilization is important in FXR-mediated iNOS and IL-1β trans-repression [9]. Thus, NF-κB signaling inhibition seems to be important in the anti-inflammatory effects of bile acid receptors and seems to occur through various mechanisms depending on the cell type and disease (Figure 2).
Therapeutic applications of bile acid receptors in chronic inflammatory diseases
Following the animal studies demonstrating the anti-inflammatory effects of FXR, phase 1 and 2 clinical trials studying FXR agonists such as INT-747 (obeticholic acid), UDCA, and CDCA have begun and are in progress in various inflammatory disorders such as primary sclerosing cholangitis, alcoholic hepatitis, primary biliary cirrhosis, type 2 diabetes, and non-alcoholic steatohepatitis. A multicenter, double-blind, placebo-controlled, parallel group, randomized clinical trial, in which obeticholic acid has been administered in non-cirrhotic, non-alcoholic steatohepatitis patients for 72 weeks, has shown that obeticholic acid is superior to placebo in improving biochemical and histological features of steatohepatitis [174748].
The FXR/TGR5 dual agonist INT-767 was superior in inhibiting liver damage compared to INT-747 (FXR agonist) or INT-777 (TGR5 agonist) alone in mouse models of sclerosing cholangitis (Mdr2-/- mice), and was also superior in inhibiting atherosclerosis in animal models (ApoE-/- mice, LDLR-/- mice) compared to non-dual agonists. This suggests that FXR/TGR5 dual agonists may be effective in the treatment of other inflammatory disorders [4349].
Although the potential for bile acid receptors as therapeutic target molecules has been studied in various inflammatory disorders, no such studies have been conducted in rheumatic disorders, including rheumatoid arthritis.
CONCLUSION
Up to now, research has revealed the anti-inflammatory and immunomodulatory effects of bile acid receptors, which is dependent on NF-κB inhibition, and the potential for bile acid receptor agonists as candidate therapeutic agents in rheumatic diseases. Although bile acid receptor-mediated anti-inflammatory effects have been seen in inflammatory disorders such as alcoholic hepatitis, non-alcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, IBDs, acute lung injury, asthma, and multiple sclerosis, most of the evidence comes from rodent studies and not studies of human cells; furthermore, studies in rheumatic disorders such as rheumatoid arthritis are also lacking. Therefore, to exploit the bile acid receptor as a novel therapeutic target in rheumatic disorders, further studies of bile acid receptor expression, function, and regulation in rheumatic disorders is warranted.
 XML Download
XML Download