

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
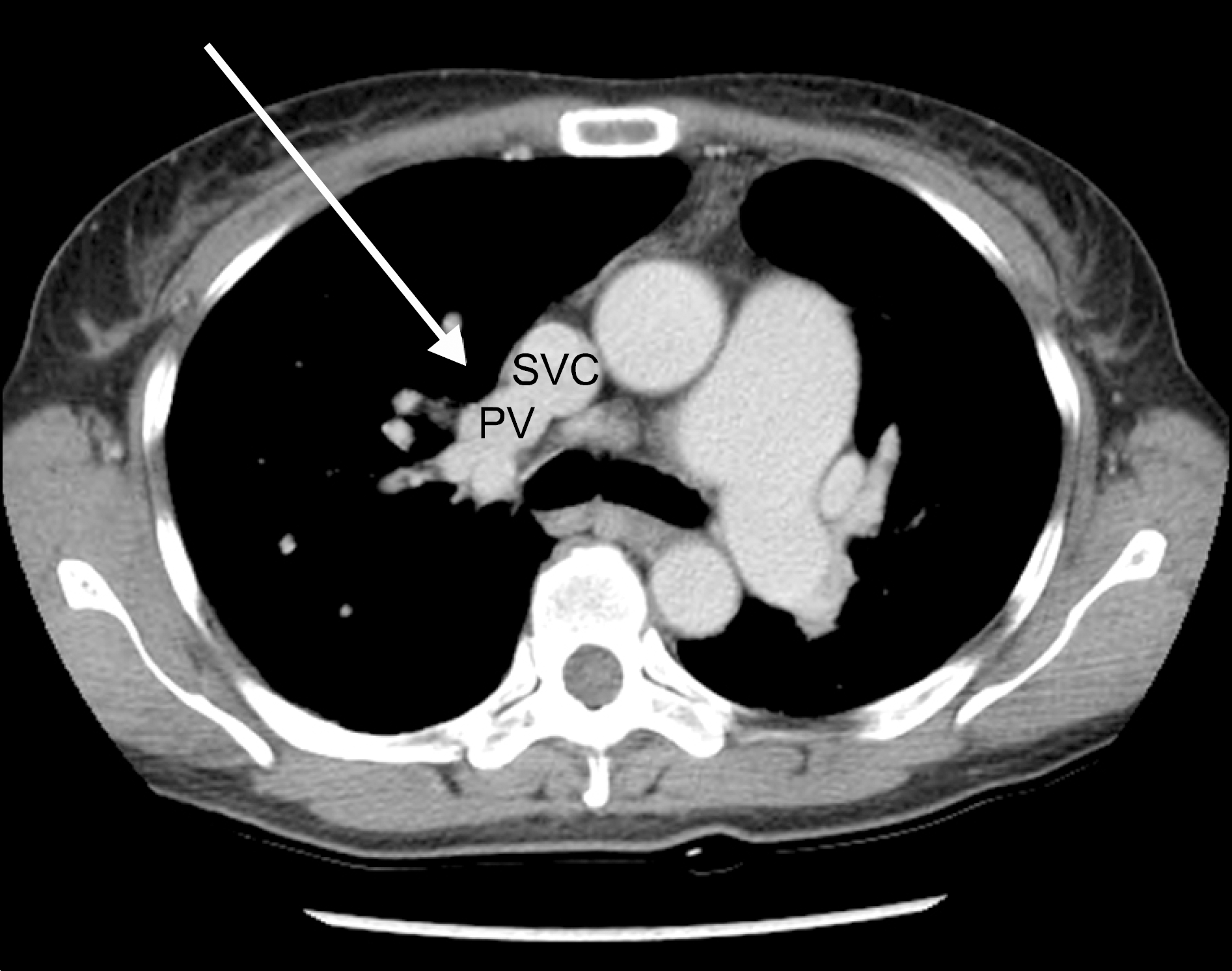
Pulmonary hypertension (PH) is a rare manifestation in patients with primary Sjogren's syndrome (pSS) and it can oc-cur with or without interstitial lung disease (ILD). Patients with PH and ILD who show signs of exacerbation of dyspnea are commonly assessed for pure PH aggravation, ILD progression or pulmonary infection. However, the presence of congenital cardiac anomalies, such as partial anomalous pulmonary vein return (PAPVR), can also be a cause of dyspnea exacerbation. PAPVR is a rare congenital anomaly that involves drainage of 1 to 3 pulmonary veins into the right-sided heart circulation, resulting in a partial left-to-right shunt. Here we present a case of PAPVR as the cause of PH aggravation in a patient with pSS with accom-panying PH.
References
1. Vachiéry JL, Simonneau G. Management of severe pulmonary arterial hypertension. Eur Respir Rev. 2010; 19:279–87.
2. Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009; 54(1 Suppl):S43–54.

3. Hoeper MM. Pulmonary hypertension in collagen vascular disease. Eur Respir J. 2002; 19:571–6.
4. Chang B, Wigley FM, White B, Wise RA. Scleroderma patients with combined pulmonary hypertension and interstitial lung disease. J Rheumatol. 2003; 30:2398–405.
5. Lei YX, Zhang X, Cui Y, Dong GF, Luo RQ. Clinical analysis of 79 pulmonary arterial hypertension cases from 1892 connective tissue disease patients. Zhonghua Yi Xue Za Zhi. 2009; 89:2934–7.
6. Ryu JH, Krowka MJ, Pellikka PA, Swanson KL, McGoon MD. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc. 2007; 82:342–50.
7. Ho ML, Bhalla S, Bierhals A, Gutierrez F. MDCT of partial anomalous pulmonary venous return (PAPVR) in adults. J Thorac Imaging. 2009; 24:89–95.
8. Launay D, Hachulla E, Hatron PY, Jais X, Simonneau G, Humbert M. Pulmonary arterial hypertension: a rare com-plication of primary Sjögren syndrome: report of 9 new cases and review of the literature. Medicine (Baltimore). 2007; 86:299–315.
9. García-Carrasco M, Ramos-Casals M, Rosas J, Pallarés L, Calvo-Alen J, Cervera R, et al. Primary Sjögren syndrome: clinical and immunologic disease patterns in a cohort of 400 patients. Medicine (Baltimore). 2002; 81:270–80.
10. Kokosi M, Riemer EC, Highland KB. Pulmonary involvement in Sjögren syndrome. Clin Chest Med. 2010; 31:489–500.
11. McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, et al. American College of Chest Physicians. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004; 126(1 Suppl):14S–34S.
12. Sanchez O, Sitbon O, Jaïs X, Simonneau G, Humbert M. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. Chest. 2006; 130:182–9.
13. Shapiro S. Management of pulmonary hypertension resulting from interstitial lung disease. Curr Opin Pulm Med. 2003; 9:426–30.
14. Toyoshima M, Sato A, Fukumoto Y, Taniguchi M, Imokawa S, Takayama S, et al. Partial anomalous pulmonary venous return showing anomalous venous return to the azygos vein. Intern Med. 1992; 31:1112–6.
 XML Download
XML Download