

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ZAKI-4 belongs to a family of proteins functioning as calcineurin regulators
ZAKI-4 gene was originally identified by us as a thyroid hormone (TH) responsive gene which locates on human chromosome 6[1]. We later found that three transcripts, α, β1 and β2, were generated by the single ZAKI-4 gene through differential splicing, and the expression of only α isoform is upregulated by TH[2]. The β1 and β2 isoforms encode the same protein product ZAKI-4β, which shares the common carboxyl terminal region with ZAKI-4α. Both ZAKI-4α and β belong to a family of small structurally related proteins which bind to, and down-regulate the activity of calcineurin (protein phosphatase 2B)[3,4]. In human other two proteins are also included in this family, DSCR1 and DSCR1L2. Recently the proteins of this family are attracting more attention since they are likely to have important functions through regulating calcineurin activity which is fundamental to so many biological processes. It was thus proposed to rename the family as RCAN (regulators of calcineurin) and their protein products as RCANs.
ZAKI-4α and calcineurin are colocalized in the brain
As we previously reported both ZAKI-4α and β bind and inhibit calcineurin activity through the common carboxyl terminus[2]. Moreover, physiological concentration of TH reduced the endogenous calcineurin activity through inducing ZAKI-4α expression. Since the tissue expression of ZAKI-4α is mainly in the brain where calcineurin is expressed abundantly, we compared the distribution of both in the brain.
Calcineurin is especially concentrated in the hippocampal and striatal neurons[5]. Subcellularly, it is present in cell bodies, postsynaptic densities, dendrites, axons, spines, and presynaptic terminals[5]. Cheung and colleagues measured the subcellular distribution of calcineurin in chick forebrain homogenate[6]. Their study showed that calcineurin was highly enriched in the cytoplasmic and microsomal fractions as well as in the synaptosomes. Although the presence of calcineurin was demonstrated in primary culture of cerebellum neurons obtained from 20-day-old rat fetuses[7], Western blot analysis of the whole brain extract from rat detected no expression of calcineurin on embryonic day 18 but it accumulated rapidly between day 4 and 5 weeks postnatally, after which it was maintained at high level[8].
We demonstrated that ZAKI-4 gene is widely expressed in neurons throughout the rat brain by in situ hybridization using a probe common to α and β[9]. Furthermore, its expression was positively regulated by thyroid hormone in certain brain areas[9]. Developmentally, its expression was detected on embryonic day 18 and increased gradually reaching a plateau at postnatal day 7. As shown in Table 1, distribution of ZAKI-4 mRNA in adult rat overlapped with that of calcineurin as detected by in situ hybridization and immunohistochemistry in many brain areas[10].
TH regulates ZAKI-4α expression via a novel nongenomic action of TR
Since ZAKI-4α is one of the endogenous calcineurin inhibitors and may play important role in the brain, we further studied its regulation by TH. Regulation of gene expression by TH is mediated through the thyroid hormone receptor (TR), usually acting as a ligand-dependent nuclear transcription factor[11]. Liganded TR binds with its cognate cis-element (thyroid hormone-responsive element, TRE) present in the regulatory region of target genes, and promotes their transcription. This TH action is often referred to as genomic action. However, we found that TH-dependent expression of ZAKI-4α is not mediated by such genomic action, because there is no canonical TRE in the promoter of ZAKI-4α gene, and because a protein synthesis inhibitor cycloheximide (CHX) abrogated TH-dependent ZAKI-4α expression, suggesting that de novo protein synthesis is required[1].
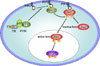
Intriguingly, TH-induced ZAKI-4α expression was inhibited by series of inhibitors, including LY294002 (inhibitor of PI3K) and rapamycin (the specific inhibitor of mammalian target of rapamycin, mTOR), suggesting the involvement of PI3K-Akt and mTOR signaling cascade[12]. Indeed, we found that 1) TH treatment induced sequential phosphorylation and activation of the serine/threonine kinase Akt, the mammalian target of rapamycin (mTOR) and its substrate p70S6K and 2) phosphorylation of these proteins was abrogated by both PI3K inhibitors, LY294002 and wortmannin, and by a dominant negative PI3K (△p85α). This TH action is very rapid and independent of protein synthesis, suggesting that it does not involve the typical nuclear action of TR. Moreover this rapid TH action was abrogated by the introduction of a dominant negative TRβ (TRG345R) into the cells expressing the wild type TRβ, suggesting the participation of TRβ.
How does TH initiate the activation of PI3K via TR? The interaction of p85α, the regulatory subunit of PI3K with other cellular proteins, including insulin receptor, insulin receptor substrate, and several members of the Rho family, has been well documented to activate PI3K activity[13~15]. For instance, binding of insulin to the membrane receptor results in the phosphorylation of its receptor and insulin receptor substrate (IRS) proteins. These proteins then interact with Src homology 2 (SH2) domain-containing proteins such as p85α, the regulatory subunit of PI3K[16]. PI3K is subsequently recruited to the membrane where it converts lipid phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], to phosphatidylinositol-3,4,5-triphosphate [PtdIns(3,4,5)P3]. The synthesis of PtdIns(3,4,5)P3 recruits the proteins possessing pleckstrin homology domain, such as Akt/PKB and phosphoinositide-dependent protein kinase 1, from the cytoplasm to the plasma membrane. Then, phosphorylation and activation of Akt/PKB occur near the plasma membrane. We thus investigated the possibility of TRβ binding to p85α. A direct interaction between TRβ and PI3K was demonstrated in human skin fibroblasts by coimmunoprecipitation of TRβ with the p85α subunit of PI3K[12]. The interaction is independent of ligand binding, while PI3K can be activated only in the presence of TH. The interaction between TRβ and p85α most likely takes place in the cytoplasm, because within minutes after activation by TH, phosphorylated Akt is translocated from the cytosol into the nucleus, where it further phosphorylates mTOR (Fig. 1). Recently, in thyroid tissue extracts, Furuya et al. demonstrated the binding between TRβ and p85α in both cytoplasm and nucleus[17], suggesting that the interaction might occur in a tissue or cell-type specific manner. The binding domain of TRβ with p85α is not clear yet, although Furuya et al suggested it within ligand binding domain. Further study is still required to clarify the interaction between TR and p85α.
Other genes except for ZAKI-4α could also be regulated by the novel non-genomic action of TH
As described above, TH induces ZAKI-4α expression in a CHX sensitive manner, suggesting ZAKI-4α as an indirectly induced gene downstream of nongenomic action of TH. The transcription factor linking PI3K pathway to ZAKI-4α expression has not yet been identified. Recently, this TH nongenomic action was reported to directly upregulate the transcription factor subunit hypoxia-inducible factor (HIF)-1α[18]. Blocking PI3K activity by inhibitors such as LY294002 and wortmannin, abrogated HIF-1α induction and CHX pretreatment did not inhibit HIF-1α mRNA increase after T3 treatment. The increase of HIF-1α results in the transcription of the target genes, including glucose transporter 1 (GLUT1), platelet-type phosphofructokinase (PFKP), and the monocarboxylate transporter 4 (MCT4). These genes are functionally related as they have important roles in cellular glucose metabolism, from glucose uptake (GLUT1) to glycolysis (PFKP) and lactate export (MCT4).
TH may play physiological or pathological roles through regulating PI3K-Akt cascade
TH plays critical roles in differentiation, growth, and metabolism. Especially in central nervous system, neonatal hypothyroidism results in severe functional deficits of the brain, including mental retardation, ataxia, spasticity and deafness. The actions of TH in central nervous system were considered to be mainly mediated by nuclear TR which regulates the transcription of target genes after T3 binding. On the other hand, nongenomic action of thyroid hormone has recently been recognized at the molecular levels, such as control of Ca2+entry, intracellular protein trafficking, and regulation of protein kinase C, mitogen-activated protein kinases and cytoskeleton[19]. However it is still not clear how they are brought about or what their physiological roles are. Recently, the extracellular portion of a transmembrane glycoprotein integrin αVβ3 was identified as a cell surface receptor for thyroid hormone [L-T4 (T4)] and as the initiation site for T4-induced activation of MAPK and subsequent proangiogenic action[20]. Our finding for the first time demonstrated a nongenomic action of TH which requires TRβ bound to p85α and causes a rapid activation of PI3K-Akt/PKB and mTOR-p70S6 kinase cascade. This leads to the expression of ZAKI-4α, an endogenous calcineurin inhibitor. TH thus may play the role in neuronal plasticity through regulating calcineurin activity. Moreover, TH-induced activation of PI3K-Akt may also play a role in regulating glucose metabolism through control the expression of other genes, such as HIF-1α GLUT1, PFKP and MCT4.
A recent study using TRβPV/PV mouse which harbors a knock-in mutant TRβ gene (PV mutant) and spontaneously develops thyroid cancer, elucidated the important role played by this nongenomic action during thyroid carcinogenesis[17]. The PV mutant has a C-insertion at cordon 448 that produces a frame shift in the C-terminal 14 amino acids of wild type TRβ[21]. It completely looses T3 binding and exhibits potent dominant-negative activity[22]. It was shown that in thyroid tumors, PV mutant bound significantly more to the PI3K-regulatory subunit p85α, resulting in a greater increase in the kinase activity contributing to thyroid carcinogenesis. Many other TRβ mutations have been found in the patients with resistance to thyroid hormone disease. To address whether those mutant TRs play pathological roles through impaired activation of PI3K-Akt, further investigation is still required.
Conclusions and prospects
TR is one of the nuclear steroid hormone receptors, which also includes estrogen receptor, glucocorticoid receptor, progesterone receptor, retinoic acid receptor and so on. As shown for estrogen receptor[23~26], TR also is involved in stimulating PI3K-Akt signaling cascade through binding with p85α. This novel finding broadened the functions of TR beyond being a nuclear transcription factor. It raises a new issue that PI3K-Akt activation could be common to all the nuclear steroid hormone families. Moreover, the novel nongenomic action of TH also promotes us to speculate that more TH responsive genes are under the control of this kinase signaling cascade, because the signal from kinase to kinase is potentially amplified while the amplification depended on TRE in the promoter region of the target genes is limited.

 XML Download
XML Download