

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Von Hippel-Lindau (VHL) disease is an autosomal dominant disease, which forms hypervascular tumors in multiple organs, such as hemangioblastomas in the retina and central nervous system, renal cell carcinomas, pheochromocytomas and cysts in various organs. Recent advances in gene testing have made it possible to screen family members for VHL disease. We experienced a 28 year-old male, who was diagnosed with bilateral pheochromocytomas through a family screening test when his elder monozygous twin brother was diagnosed with a pheochromocytoma. He received no treatment until December, 2004, when he visited the Emergency room due to a headache. A hemangioma of the cerebellum was seen in the brain MR study, leading to the diagnosis of type 2A VHL disease. An abdominal CT scan revealed no lesions of the pancreas or kidney. There was no evidence of a hemangioma in the retinal scan. The subsequent gene testing showed a germline mutation in exon 3 codon 167 of the VHL gene. The mother of the patient was revealed to have the same mutation of the VHL gene, but the elder brother of the patient did not.
Figures and Tables
Fig. 1
Germline mutation of the VHL gene. A missence mutation (C to T transversion) at codon 160 on short arm of chromosome 3 was detected, which leads to an amino acid change from arginine to tryptopane. N indicates C >T change in mutant allele

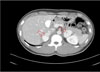
Fig. 2
Abdominal CT. Rt. 3.7 cm and Lt. 3.8 cm sized contrast enhancing solid mass lesion are noted on both adrenal gland

References
1. Richard S, Graff J, Lindau J, Resche F. Von Hippel-Lindau disease. Lancet. 2004. 363:1231–1234.
2. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003. 361:2059–2067.
11. Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: The expanding genetic differential diagnosis. J Nat Cancer Inst. 2003. 95:1196–1204.
12. Richards FM, Webster AR, Mcmahon R, Woodward ER, Rose S, Maher ER. Molecular genetic analysis of von Hippel-Lindau disease. J Intern Med. 1998. 243:527–533.
13. von Hippel E. Die anatomische Grundlage der von mir beschriebenen "sehr seltene Erkrankung der Netzhaut". A von Graefe's Arch Ophthalmo. 1911. 79:350–377.
14. Brandt R. Zur Frage der Angiomatosis retinae. A von Graefe's Arch Ophthalmol. 1921. 106:127–136.
15. Lindau A. Zur Frage der Angiomatosis retinae und ihrer Hirnkomplikationen. Acta Ophthalmol (Copenh). 1927. 4:193–226.
16. Lindau A, Sargent P, Collins T, Greenfield JG, Riddoch G, Russell D, Rawling LB, Weber FP, Cohen H, Barnard WG. Discussion on vascular tumours of the brain and spinal cord. Proc R Soc Med. 1931. 24:363–370.
17. Maher ER, Kaelin WG Jr. von Hippel-Lindau disease. Medicine(Baltimore). 1997. 76:381–391.
 XML Download
XML Download