

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ozone is well known as an important component of ambient air pollutants. Exposure of ozone to patients with bronchial asthma can aggravate asthmatic symptoms and increase hospital admission due to exacerbation of bronchial asthma [1]. The visit of Emergency Department associated with maximal daily 1- and 8-hour average ozone concentration in children [2]. In adult, also, long-term exposure to ambient ozone was associated with the development of bronchial asthma [3].
Patients with bronchial asthma have increased susceptibility to ozone because ozone has induced aggravation of underlying allergic airways inflammation. These inflammatory reactions were evidenced by increased percentages of eosinophils [45] and increased exhaled nitric oxide levels [6] after ozone exposure. In healthy person, also, ozone exposure induces significant decrement of lung function and neutrophilic inflammation of airway [7] instead of eosinophilic inflammation as shown in asthmatics. In regard to oxidative system, in healthy person, increased concentration of total glutathione and extracellular superoxide dismutase occurred 6 hours post ozone exposure in bronchoalveolar lavage (BAL) fluid, suggesting ozone elicits airway antioxidant responses [8]. To our knowledge there is little research regarding oxidative system after ozone exposure in asthmatics.
The response to ozone increased airway inflammation in atopic asthmatics compared to nonatopic asthmatics [9], and atopic subjects with or without bronchial asthma showed association of increasing ozone exposure with lower lung function and increased biomarkers of respiratory and systemic inflammation [6]. These findings suggested atopic status itself may be important factor in generation of ozone-induced inflammatory airway responses. Ozone can aggravate respiratory symptoms in patients with bronchial asthma, but, not in healthy person.
It has been known that increase of oxidative stress and derangements of antioxidant system in asthmatic airway [10]. But, there was no report regarding oxidative stress and antioxidant activity after ozone exposure in asthmatic and nonasthmatic airway. We hypothesized asthma itself may show different response to ozone, as atopic status make difference response to ozone. This study was performed to evaluate the differences of response to ozone between normal mice and asthmatic mice model in terms of status of oxidant injury and antioxidant activity.
Go to : 
MATERIALS AND METHODS
Animals and experimental design
Experiments protocols were performed according to the Guidelines for the Care and Use of Laboratory Animals of the National Health Institute. Female BALB/c mice 5–6 weeks of age were obtained from Daehan Laboratories (Daejeon, Korea). The mice were maintained on ovalbumin (OVA) free diets. The mice were individually housed in rack-mounted stainless steel cages with free access to food and water.
Ozone exposure and the measurement of airway responsiveness were performed on the 24, 27, 31, 38, and 45 experimental days. Airway responsiveness was measured after ozone exposure and BAL was performed immediately after the last measurement of airway responsiveness (Fig. 1). Following BAL, the chest wall was opened. The lungs were snap-frozen in liquid nitrogen and stored for Western blot and measurement of glutathione S-transferase (GST) activity.
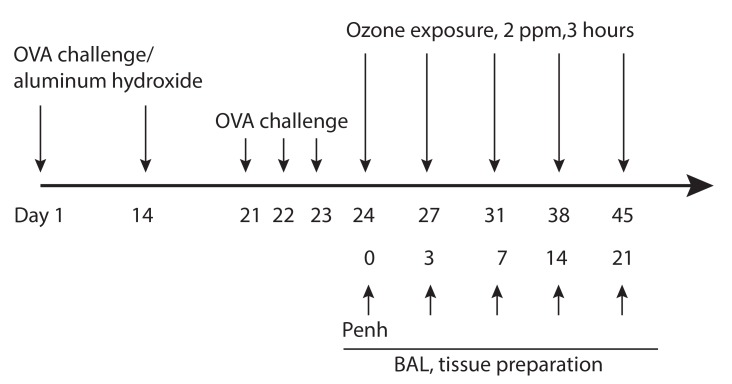 | Fig. 1Schematic diagram of the experimental protocol. Mice were sensitized on day 1 and 14 by intraperitoneal injection of ovalbumin (OVA) with aluminum sulfate. On day 21, 22, and 23 after the initial sensitization, the mice were challenged with an aerosol of 1% OVA using ultrasonic nebulizer. The mice housed in whole-body exposure chambers were exposed to ozone concentration of 2 ppm for 3 hours on day 24, 27, 31, 38, and 45 after initial OVA sensitization. ppm, parts per million; Penh, enhanced pause; BAL,bronchoalveolar lavage.
|
OVA-induced asthma model
An OVA-induced asthma model (OVA-model, n = 6) was constructed with modified method previously described [11]. Briefly, mice were sensitized by means of intraperitoneal injection of 10 g of grade V OVA (Sigma Chemical, St Louis, MO, USA) and 1 mg of aluminum potassium sulfate (Sigma Chemical) in saline solution on day 1 and 14 days. Vehicle control mice (sham model, n = 6) were sensitized with a suspension of aluminum potassium sulfate (1 mg) in saline solution. Each group was challenged with 5% OVA or saline solution daily from days 21 to 23 (Fig. 1). Aerosol challenge was carried out on groups of up 20 mice in a closed system chamber attached to an ultrasonic nebulizer (NE-UO7; Omron Corp., Tokyo, Japan) with an output of 1 mL/min and 1- to 5-µm particle size.
Determination of airway responsiveness to methacholine
Airway responsiveness was measured in unrestrained and conscious mice as previously described [11]. Mice were placed in a barometric plethysmographic chamber (All Medicus Co., Seoul, Korea), and baseline readings were taken and averaged for 3 minutes. Aerosolized methacholine in increasing concentrations (from 2.5 to 100 mg/mL) were nebulized through an inlet of the main chamber for 3 minutes. Readings were taken and averaged for 3 minutes after each nebulization and enhanced pause (Penh) was determined. Penh, calculated as (expiratory time/relaxation time-1)×(peak expiratory flow/peak inspiratory flow) according to the manufacturers' protocol, is a dimensionless value that represents a function of the proportion of maximal expiratory to maximal inspiratory box pressure signals and a function of the timing of expiration. Results are expressed as the percentage increase of Penh following challenge with each concentration of methacholine, where the baseline Penh (after saline challenge) is expressed as 100%. Penh values averaged for 3 minutes after each nebulization were evaluated.
Ozone exposure
The mice housed in whole-body exposure chambers were exposed to ozone concentrations of 2 parts per million (ppm) for 3 hours after OVA challenge on days 24 to 45 (Fig. 1). Ozone was generated with Sander Model 50 ozonizers (Sander, Eltze, Germany). The concentration of ozone within the chambers was monitored throughout the exposure with ambient-air ozone motors (Model 49C; Thermo Environmental Instruments Inc., Franklin, MA, USA). The air-sampling probes were placed in the breathing zone of the mice. The mean chamber ozone concentration (±standard error of the mean) during the 3-hour exposure period was 1.98 ± 0.08 ppm. Temperature and humidity were maintained at a constant level within the chamber.
BAL fluid preparation and analysis
The mice were deeply anesthetized by the use of 50 mg/kg of pentobarbital sodium via the intraperitoneal injection and were killed by exsanguination from the abdominal aorta. The trachea was cannulated with a polyethylene tube through which the lungs were lavaged four times with 1.0 mL of physiologic saline (4.0 mL total). The BAL fluid was filtered through wet 4 × 4 gauze. Trypan blue exclusion for viability and total cell count were performed. The BAL fluid was centrifuged at 150 × g for 10 minures. The obtained pellet was immediately suspended in 4 mL of physiologic saline, and total cell numbers in the BAL fluid were counted in duplicate with hemocytometer (improved Neubauer counting chamber). Then, a 100-L aliquot was centrifuged in a cytocentrifuge (Model 2 Cytospin; Shandon Scientific Co., Pittsburg, PA, USA). Differential cell counts were made from centrifuged preparations stained with Diff-quick, counting 500 or more cells in each animal at a magnification × 1,000 (oil immersion).
Western blot
First, 20 µg of protein were electrophoresed in a 15% polyacrylamide gel with a discontinuous system. The proteins were transferred to a nitrocellulose membrane at 120 V for 40 minutes. After blocking in 5% skim milk and 0.1% NP40 in Trisbuffered saline for 2 hours at room temperature, membranes were incubated with different primary antibodies. Mouse antimouse antibody against PCNA, HNA (Abcam, Cambridge, MA, USA), and rabbit anti-mouse against NF-E2 related factor 2 (Nrf-2) were utilized. The membrane was incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (1:5,000 dilution) for 1 hour at room temperature. The target protein was detected using enhanced chemiluminescence solution (Amersham Pharmacia Biotech, Little Chalfont, UK).
Measurement of GST activity
GST activity in tissue homogenates were measured according to the manufacture's protocol (Cayman Chemical Co., Ann Arbor, MI, USA) which measuring the conjugation of 1-chloro-2,4-dinitrobenzene reducing glutathione.
Statistical analysis
Mann-Whitney U-test was used to compare the differences of Penh, cellular pattern in BAL fluid, and GST activity between OVA-model and sham model. All statistical analyses were performed using SPSS ver. 14.0 (SPSS Inc., Armonk, NY, USA). A p < 0.05 was considered to be statistically significant.
Go to :
RESULTS
Sensitization and BAL fluid analysis
OVA-induced asthma model (OVA-model) had a higher methacholine-induced Penh compared to those in the sham model at 25 and 50 mg/mL of methacholine concentration (p < 0.01) (Fig. 2).

Total cell count gradually decreased from basal state (before ozone exposure) to 21 days in OVA-model, but there was no change in sham model. Total cell count significantly increased in OVA-model compared to those in sham model during whole experimental period (Fig. 3A). The change of macrophage showed similar pattern with total cell count (Fig. 3B). The numbers of lymphocyte significantly increased in OVA-model compared to those in sham model. The change of lymphocyte gradually decreased from basal state to 21 days except 14 days after ozone exposure in OVA-model (Fig. 3C). Eosinophils significantly increased at basal state, 3 and 7 days after ozone exposure in OVA-model compared to those in sham model. There were no difference at 14 and 21 days between 2 groups (Fig. 3D). The numbers of neutrophil were too small compared to other cells and were not different between 2 groups (data are not shown).
 | Fig. 3(A) Total cell count and inflammatory cell counts in bronchoalveolar lavage fluid from the ovalbumin (OVA)-induced asthma (blue bar) and sham model (green bar). Total cell count significantly increased in OVA-induced asthma model compared to those in sham model during whole experimental period. (B, C) The numbers of macrophage and lymphocyte showed significantly increased in OVA-induced asthma model compared to those in sham model. (D) Eosinophils significantly increased at basal state, 3 and 7 days after ozone exposure in OVA-induced asthma model compared to those in sham model. (A) *p < 0.01 vs. 7, 14, and 21 days, #p < 0.01 vs. 14 and 21 days, ¶p < 0.01 vs. 21 days, $p < 0.05 vs. control. (B) *p < 0.01 vs. 14 and 21 days, $p < 0.05 vs. control. (C) *p < 0.01 vs. 21 days, #p < 0.05 vs. 14 and 21 days, $p < 0.01 vs. control. (D) *p < 0.01 vs. 7, 14, and 21 days, #p < 0.01 vs. 14 and 21 days, $p < 0.01 vs. control.
|
Expression of 4-hydroxy-2-nonenal
To know lipid peroxidation by ozone, we measured protein expression of 4–HNE with Western blot. In sham model, the expression of 4-hydroxy-2-nonenal (4-HNE) increased at only 14 days during experimental period. In OVA-model, the expression of 4-HNE already more increased at baseline (without ozone) compared to those in sham model. This increased expression is more enhanced at 3 days after ozone exposure. At 7 days after ozone exposure the expression decreased to baseline level and at 14 and 21 days after ozone exposure the expression is significantly decreased below the level of baseline (Fig. 4A).
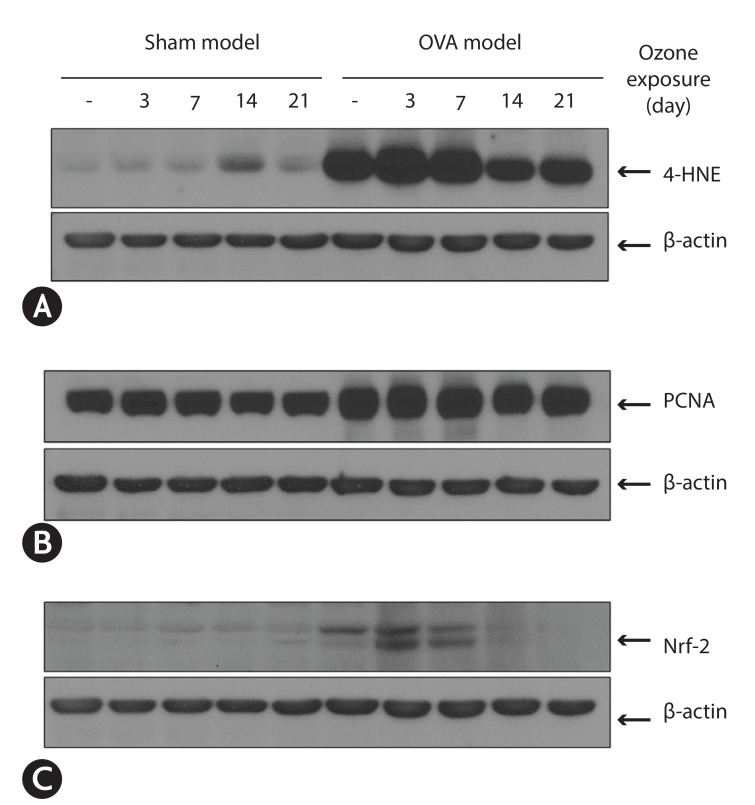 | Fig. 4Western blot analysis of 4-HNE (A), PCNA (B), and Nrf-2 (C) after exposure to ozone in ovalbumin (OVA)-induced asthma and sham-model. (A) In OVA-induced asthma model, the expression of 4-HNE already more increased at baseline compared to those in sham model. This increased expression is more enhanced at 3 days after ozone exposure. At 7 days after ozone exposure the expression decreased to baseline level and at 14 and 21 days after ozone exposure the expression is significantly decreased below the level of baseline. (B) In sham and OVA-induced asthma model, PCNA expressions were not different from baseline to 21 days. But the expression of PCNA was significantly increased in OVA-model compared to those in sham model. (C) In sham model, the expression of Nrf-2 was not observed at baseline and during experimental period. In contrast, in OVA-model, expression of Nrf-2 was observed at baseline, and 3 and 7 days after exposure ozone. OVA; ovalbumin, 4-HNE; 4-hydroxy-2-nonenal, PCNA; proliferative cellular nuclear antigen, Nrf-2; NF-E2-relatd factor 2.
|
Expression of proliferating cell nuclear antigen
The cellular proliferation, the result of lipid peroxidation, was measured with proliferating cell nuclear antigen (PCNA) expression by Western blot. In sham and OVA-induced asthma model, PCNA expressions were not different from baseline to 21 days. But the expression of PCNA was significantly increased in OVA-model compared to those in sham model (Fig. 4B).
Western blot of Nrf-2
To know the defense mechanisms of oxidative stress, we measured the level of Nrf-2 by Western blot. In sham model, the expression of Nrf-2 was not observed at baseline and during experimental period. In contrast, in OVA-model, expression of Nrf-2 was observed at baseline, and 3 and 7 days after exposure ozone. At 14 and 21 days after ozone treatment expression of NRf-2 was not observed (Fig. 4C).
GST activity
The activity of GST was measured with protein from whole lung lysate. In sham model, the level of GST activity was not different during experimental periods, but, in OVA-model, the activity of GST increased significantly after exposure of ozone (Fig. 5).
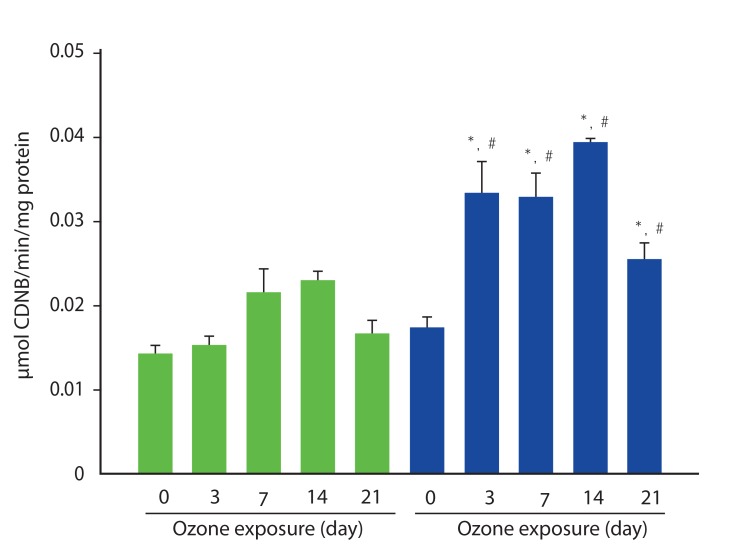 | Fig. 5Glutathione activity of lung from ovalbumin (OVA)-induced asthma (blue bar) and sham-model (green bar). The activity of glutathione was more increased in OVA-induced asthma model compared to that in sham model. The exposure to ozone significantly increased glutathione activity in OVA-induced sham model. *p < 0.05 vs. sham model, #p < 0.05 vs. 0 day. CDNB, 1-Chloro-2,4-dinitrobenzene.
|
Go to :
DISCUSSION
In present study, asthmatic airway and nonasthmatic airway showed different responses to ozone. Asthmatic airway showed elevated level of 4-HNE (oxidative stress), Nrf-2, and glutathione (antioxidant activity) unlike nonasthmatic airway which did not show the elevation of above proteins after exposure to ozone. We expected asthmatic airway showed more exaggerated oxidative stress and more decreased antioxidant activity compared to nonasthmatic airway after ozone exposure. But, unexpectedly, antioxidant activity was more prominent in asthmatic airway compared to that in nonasthmatic airway. The possible reasons to explain these phenomena are as follows; first, the ozone concentration used in the present study was lower than threshold which evokes inflammation in nonasthmatic airway. The exposure of 0.06-ppm ozone for 6.6 hours causes significant increase of neutrophilic inflammation in the airways in healthy young adults [7]. Although 0.06-ppm ozone concentration was much lower than that of present study, they stimulated airway with exercise and zone together [7]. Therefore, we do not know yet the exact concentration of ozone to evoke airway inflammation. Second, the airway of the animal asthma model could not be the same as airway of patients with bronchial asthma. Human asthma is associated with acute and chronic inflammation of the airway mucosa, in contrast, experimental asthma models shows acute allergic inflammation associated with alveolitis [12]. Third, there was no report regarding the change of antioxidant activity after ozone exposure in patients with bronchial asthma. Although increasing ambient ozone exposure predicted lower lung function and increased respiratory inflammation, they did not measure oxidative system including antioxidants activity [6]. Therefore, we could not exclude the possibility of increasing antioxidant activity after exposure of ozone in patients with bronchial asthma in real fields.
4-HNE, result of lipid peroxidation, was used as an indicator of ozone responses in airway, because of 4-HNE after ozone exposure has a potential role in the cellular toxic effect in airway [13]. The expression of 4-HNE was totally different between sham- and OVA-model. In sham-model, 4-HNE slightly expressed only at 14 days after ozone exposure, but in OVA-model, 4-HNE spontaneously expressed from baseline (after ovalbumin challenge). In experiment with murine asthma model using ovalbumin, OVA-sensitization induced a modest increase of 4-HNE in immunofluorescence study [14] and malondialdehyde, one of oxidant parameter, increased significantly in the patients with acute exacerbation of asthma compared to those in patients with stable asthma [15]. These findings are consistent with our results. Because of limited data regarding measuring 4-HNE in patients with asthma or murine asthma model, it is very hard to explain why OVA-sensitization increase the level of 4-HNE. This phenomenon may be explained by the relationship with increased expression of 4-HNE and eosinophilic inflammation in atopic cornea [16]. They showed the positive correlation with the percentage of eosinophils and the expression of 4-HNE. We observed, in OVA-model, BAL fluid showed elevated numbers of eosinophils from baseline to 7 days after ozone exposure which were well correlated with the expression of 4-HNE in Western blot. 4-HNE mainly expressed in bronchial epithelium after ozone exposure in OVA-model. These findings were also observed in mice under oxidative stress [17], suggesting present experiment well express the oxygen toxicity in OVA-induced animal asthma model.
We measured PCNA expression, the response of lipid peroxidation, to know oxygen toxicity affecting bronchiolar epithelium. The expression of PCNA more increased in OVA-model compared to that in sham model after ozone exposure. This pattern was similar with the pattern of 4-NHE after ozone exposure. In sham model, the expression of PCNA was observed before ozone exposure. This finding was consistent with other study showing PCNA expression in lung of mice before ozone exposure [18].
Nrf-2 has a crucial role in the induction of antioxidant response element which up-regulates expression of several antioxidant gene [19]. In this study, interestingly, Nrf-2 increased in OVA-model not in sham model. These findings suggest the protective activity from oxidant injury was more increased in asthma model in spite of airway inflammation. Furthermore, it is very interesting to find out elevated GST activity only in OVA-model not in sham model. We speculated the increased activity of GST may detoxify ozone-induced oxygen toxicity. The role of GST has been known as detoxification of oxygen toxicity action of 4-HNE by converting 4-HNE to GS-4-hydroxy-2-nonenal by GST [20]. Antioxidant capacity was lower in patients with bronchial asthma as compared to controls and further decreased in asthmatics following segmental antigen challenge [21]. The discrepancy with the present study may be related with stimulator, ozone and antigen. There was no report regarding antioxidant activity after ozone exposure in patients with bronchial asthma. Therefore, the possibility of increase of antioxidant activity after ozone exposure in asthmatic airway canot be excluded.
The pitfalls of this study were as follows; first, the murine asthma model in this study do not fit choric asthma model resembling human stable bronchial asthma. Short-term exposures to ovalbumin are not likely to make chronic inflammatory and epithelial changes of airway shown in typical bronchial asthma [12]. But, we confirmed bronchial hyperreactivity and elevated eosinophils in BAL fluid in OVA-induced asthma model. These findings are accepted as the model of bronchial asthma in majority of OVA-induced murine asthma study [2223]. Second, the concentration of ozone used in the present study was totally different to that of ambient ozone and of ozone used in human study. The concentration of ozone in the present study was 2 ppm, but ambient 8-hour average ozone concentrations were 32–46.5 parts per billion [23] and, in human study, the concentration of ozone was 0.27 ppm [5]. Although the concentration of ozone causing a damage to the airway of human and mouse is different, the effects of ozone on the way are similar. It does not seem to affect the results of present study. Third, we did not measure oxidants like superoxide, hydroxyl radicals and H2O2. Because of these oxidants are highly reactive and short-lived [24], we observed the damage inflicted by these oxidants by measuring 4-HNE and PCNA. Fourth, we should include the group of ozone challenge in sham model and sham challenge of ozone in OVA-model to know the effects of OVA itself on the oxidant system. But, the cellular pattern in BAL fluid in the present study using OVA and ozone challenge is different from that in other studies [2526] using OVA challenge only. This finding suggested ozone also affects the inflammation and oxidant system. However, we could not exclude a possibility of the effect of OVA itself on inflammation and oxidant systems in the present study.
In conclusion, murine asthma model has enhanced oxygen toxicity and antioxidant activity response to ozone.
Go to :
 XML Download
XML Download