

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Drug hypersensitivity reactions (DHR) are adverse drug reactions (ADR) that are immune mediated. The term 'drug allergy' can be used for ADR that has a proven immunological mechanism and therefore overlaps with considerably drug hypersensitivity but there is a lack of consensus on its exact definition [1]. Therefore in this document, drug hypersensitivity will be used preferentially. However, it must be noted that the term 'drug allergy' is frequently used to refer to any ADR—even by clinicians—leading to misdiagnoses in many individuals. This is particularly the case for antibiotics, and is driven by apprehension about the possibility of catastrophic anaphylaxis. This is despite true allergies accounting for less than 10% of all ADR, and of these, T-cell-mediated (i.e., delayed) reactions are by far the most prevalent [23]. Treatment thenceforth with alternate medications that may carry greater risk of toxicity, are less effective and more expensive, in turn translate to greater morbidity and economic cost.
Individual associations between particular human leukocyte antigens (HLA) and T-cell-mediated drug hypersensitivity, such as for HLA-B*15:02 with carbamazepine, HLA-B*57:01 with abacavir, and HLA-B*58:01 with allopurinol have been identified, and the availability of testing has created opportunities for optimising treatment based on personalised characteristics. The mechanisms of these reactions have been elucidated in the recent years, contributing to a greater understanding of T-cell-mediated drug hypersensitivity. The dominant model for the sensitisation of T cells by drugs has long been the hapten/pro-hapten hypothesis. In recent years this has been challenged by evidences supporting alternative pathogenic pathways, resulting in the emergence of the pharmacological interaction with immune receptors (p-i) concept and the altered peptide repertoire hypothesis.
In this review, we aim to summarise the existing concepts of T-cell-mediated drug hypersensitivity, focussing on the available experimental evidence, and possible clinical relevance.
HAPTEN/PRO-HAPTEN MECHANISM
Experiments using guinea pigs in the late 1930s showed that certain electrophilic benzene derivatives could lead to a delayed onset eczema if conjugated to the organic base aniline [45]. From this Landsteiner and Jacobs [45] deduced that simple chemicals could sensitise the immune system if they combined with an animal protein. The term coined for this immunogenic conjugation was 'haptenisation'—taken from the Greek verb for 'to fasten'. Under this model, a low molecular weight substance (typically <1,000 Daltons)—the hapten – was deemed incapable of triggering a specific immune response without binding to a much larger protein or peptide.
Although any electrophilic (and hence reactive) chemical may behave as a hapten, amongst drugs, the phenomenon of haptenation is best described for the beta-lactam antibiotics [678]. Beta-lactams display a selectivity in their target proteins, binding to extracellular proteins—particularly albumin—but not to cellular proteins [910]. It has also been demonstrated in multiple experiments that the beta-lactams have a preference for lysine residues [111213].
An implied requirement of the hapten hypothesis is that a drug must be sufficiently reactive to spontaneously bind to a carrier protein. However, many chemically inert drugs are still able to produce DHR. This phenomenon is reconciled within the hapten hypothesis by the concept of 'pro-haptens', whereby an otherwise nonreactive drug acquires reactivity by virtue of its metabolites. Sulfamethoxazole is the archetypal example of a pro-hapten, hepatically modified via the CYP2C9 isoenzyme to a pro-reactive hydroxylamine metabolite, which is in turn converted spontaneously both within and outside the liver to the toxic nitroso sulfamethoxazole. Nitroso sulphamethoxazole is unstable, and readily binds to protein residues, but unlike beta-lactams, it has a strong affinity for cysteine residues, and can haptenate both extracellular and cellular proteins [14151617].
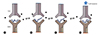
Demonstrating the existence of immune responses to haptenated proteins has proven more elusive, with the best evidence identified from a small group of individuals with cystic fibrosis, all with a clinical history of delayed reactions to the beta-lactam piperacillin [18]. From these subjects, T-cell clones (TCC) specific for piperacillin-albumin conjugates were successfully identified and cultured. These TCCs included both CD4+ helper and CD8+ cytotoxic T cells capable of recognising piperacillin-peptide conjugates presented on MHC (Fig. 1A).
In addition to these, exposing these lymphocytes to piperacillin in vitro led to measurable proliferation and activation. Interleukins (IL)-4, -5, and -13—cytokines consistent with a type 2 helper T-cell effector response—were secreted by piperacillin-specific T cells. Cytolysis of target cells was also observed. The degree of activation was also dependent upon the length of incubation with piperacillin, suggesting that the longer time was required for haptenation of albumin to occur. However, it is worth noting the heterogeneity of the T lymphocytes specific for piperacillin-albumin conjugates. Also, similar findings have since been reported for carbamazepine, a drug not known for its properties as a hapten, suggesting that the immunophenotype of T cells in drug hypersensitivity may not be specific for the type of clinical response, or even for the pathway of immune sensitisation.
Early studies aimed at uncovering the antigenic determinants of benzylpenicillin found that the penicilloyl and penicillenic acid degradation products triggered a delayed dermatitis in experimental animals [611]. However, these antigenic determinants are now also used to test for immediate hypersensitivity via skin testing protocols, suggesting that the same antigenic determinants may be capable of triggered both delayed and acute reactions.
This implies that while the hapten hypothesis provides a viable mechanism for the formation of immunogenic drug-derived neoantigens, it remains unclear how particular antigens, or the mode of haptenation might be associated with the clinical phenotype. The immunophenotyping and lymphocyte function experiments described above show that a haptenated protein could conceivably trigger both humoral or cell-mediated immune responses, leading to two very different clinical responses; the former potentially to anaphylaxis, and the latter to a delayed (T-cell-mediated) allergy. Thus the hapten/pro-hapten hypothesis provides only the beginnings of an explanation for T-cell-mediated drug allergy. It also currently remains unknown why haptenation, despite occurring as a phenomenon with drugs in most people, goes onto initiate clinical allergy in only a minority. Also unclear is the stimulation of innate immunity by haptenation, which—as discussed under danger signals— is a requirement for classical hapten specific immune responses.
IMMUNE MECHANISM OF P-I CONCEPT
Over the past two decades, it became apparent that hapten/pro-hapten concept was insufficient to explain certain findings of DHR. In particular detailed analyses of drug-specific TCC have shown that they are activated by HLA-dependent but processing- and metabolism-independent pathway [19]. Therefore p-i concept was formulated where a drug binds nonreactive to immune receptors, particularly T-cell receptors (TCR) and/or HLA [20]. The key postulates of p-i concept are: (1) the interaction occurs between a drug and TCR and/or HLA, and both HLA and TCR are required for T-cell activation; (2) the drug-receptor binding is processing- and metabolism-independent; (3) the drug binding is labile in that washing drug-pulsed antigen presenting cells (APC) do not result in T-cell activation. An exception is abacavir, which can bind in the endoplasmic reticulum to HLA-B*57:01; and (4) drug-reactive T cells are activated immediately in the presence of the drug. Only abacavir can induce reactivity in TCC after hours as abacavir is loaded inside the cells onto HLA (Table 1) [1921222324].
While it was initially thought that a drug interacts with TCR and peptide-MHC complexes (pMHC) or both (Fig. 1B, F), this is not a part of the original definition nor a proven characteristic. Rather, it was an explanation for how a drug could activate immune receptors via noncovalent binding. Subsequent further studies have shown that the mode of drug binding to TCR and/or HLA is variable; in some instances, drug binding actually occurs outside the interacting sites of TCR-pMHC (Fig. 1C, E, G) [252627282930].
There are several important features and experimental findings of p-i concept. Firstly, the interaction is generally thought to have functional consequences. In other words, a drug may bind TCR and/or HLA but if there is no ensuing T-cell response, then this would not be considered a part of p-i concept. On the other hand, a drug which does not result in T-cell response may still inhibit another structurally related compound from activating T cells by competing for the same binding site (Fig. 1D). This interesting concept was recently illustrated by showing antagonistic effect of sulfanilamides on suppressing sulfamethoxazole induced proliferation [31]. A particular sulfamethoxazole TCC named 1.3 could be activated only by sulfamethoxazole but not by other sulfanilamides. However, when TCC 1.3 was coincubated with sulfamethoxazole and another sulfanilamides, there was dose-dependent inhibition of T-cell proliferation. This implies that a certain drug may be an immune receptor antagonist whose activity can be demonstrated only in the presence of another activating drug (p-i antagonism). Therefore one could argue that this should be included in p-i concept where it may have 'beneficial' role by inhibiting unwanted T-cell activation. So far this concept has been shown only for p-i TCR (see below).
Secondly, the p-i mechanism resulting in T-cell activation is immediate with most drugs and can occur within seconds of the drug exposure [22273233]. This can only be explained by interactions between drug, TCR and pMHC that occur on the cell surface. APC that are fixed with glutaraldehyde can still activate drug-specific TCCs in the presence of the soluble drug further supporting that the interaction takes place on the cell surface [1921]. In contrast, immediate reactivity is incompatible with altered peptide repertoire as this occurs intracellularly and is processing-dependent. Hapten binding can occur on the cell surface and therefore can be rapid but it would not be reversible and washing step would not remove reactivity. Thirdly, the p-i mechanism is reversible as washing steps are often used to remove 'non-stably' bound drugs. However, drug with strong enough affinity may resist this washing step as indeed was the case for abacavir binding to HLA-B*57:01. Hapten resists the washing steps as it forms irreversible covalent bond. Finally, innate immune system is not activated and B-cell stimulation is not involved. So far there is no B-cell activation or IgE-mediated reaction described for p-i mechanism. B-cell response is unlikely as the drug interaction is limited to TCR and/or HLA and antigen processing, which is absent in p-i concept, is a critical step for adaptive response in B cells [20].
The p-i concept can be further divided into p-i TCR and p-i HLA (Fig. 1) [24]. This concept has been extensively discussed in the recent review by Pichler et al. [25]. In p-i TCR (Fig. 1B, C), a drug binds to TCR preferentially and can activate T cells via strengthening its interaction with peptide-HLA (pHLA). The drug may bind to TCR itself with high affinity at sites that makes contact with pMHC resulting in T-cell activation, even in the presence of different peptides or allogeneic HLA (Fig. 1B) [313435]. Alternatively, T-cell activation can occur via allosterism where a drug binds TCR in a distant site but alters overall TCR configuration, resulting in higher affinity for self-pMHC (Fig. 1C) [26].
In p-i HLA, a drug interacts predominantly with pMHC. In case for abacavir and allopurinol in HLA-HLA-B*57:01 and B*58:01, respectively, the drug binds to peptide binding groove [272829]. In the case of abacavir and HLA-B*57:01, alteration of self peptide repertoire additionally occurs (Fig. 1G; see below for further details). In the case of HLA-B*58:01, there is no evidence that altered peptide repertoire occurs but the binding is still likely to take place in peptide binding groove which is hidden under the peptide in the surface (Fig. 1E) [27]. Yet the reactivity pattern was consistent with p-i concept. Therefore it was proposed that peptide flexibility results in partial detachment, which then allows drug binding to occur, ultimately resulting in altered configuration of self-pHLA in the presence of the drug (Fig. 2) [27]. As summarised by Pichler et al. [25], T cells activated by p-i HLA mechanism share a number of similarity with alloreactive T cells [30]. In other words, DHR due to T-cell activation is not elicited by an antigen presented on self-HLA but rather by activation of T cells which recognises altered configuration of self-peptide present on self-HLA due to the drug binding. Whether this alloreactivity concept for DHR is relevant for any drugs stimulating via p-i HLA or p-i TCR is still unclear.
In the case of carbamazepine and HLA-B*15:02, both p-i HLA and p-i TCR appear to coexist (Fig. 1F). TCR repertoire is important as the presence of certain clonotypes were required for carbamazepine recognition [36]. On the other hand, the drug still binds to HLA-B*15:02 and residue Asn63 was shown to be critical for the drug binding [37]. Consistent with these 2 studies, a recent computational paper by Zhou et al. [38] showed that carbamazepine interacts with both TCR and HLA-B*15:02, suggesting that it exhibits the features of both p-i TCR and p-i HLA (Fig. 1F).
CLINICAL RELEVANCE OF P-I CONCEPT
There are several important clinical implications that can be derived from p-i concept. Firstly DHR with strong HLA associations typically result in severe clinical manifestations. Conversely, two of the most common causes of Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN), i.e. allopurinol and carbamazepine, are strongly associated with HLA-B*58:01 and HLA-B*15:02, respectively. Other drugs that are known to cause severe cutaneous adverse reactions (SCAR) also have various HLA associations but with lower odds ratio [39]. So far, most of HLA associated DHR appear to be driven by the p-i mechanism [27323337]. This suggests that SCAR may be predominantly driven by the p-i mechanism, more specifically p-i HLA. From drug research and development point of view, there is an argument to perform in silico analysis with modelling and docking algorithm to see if a candidate drug could bind strongly to common HLA alleles. Indeed, Yang et al. [40] predicted using in silico method that abacavir would bind HLA-B*57:01 before crystallography data became available. Such approach may be a useful additional step in assessing immunogenicity of a drug. However, it must be emphasised that such prediction must be further validated with in vitro analysis.
As mentioned earlier, p-i concept has been proven for T-cell reactivity but has not been shown for B-cell response. In support of this, most common culprits of SCAR, such as allopurinol and antiepileptic drugs, are not known to or rarely cause anaphylaxis. On the other hand, beta lactams and sulfamethoxazole can cause both maculopapular exathema (MPE) and anaphylaxis since they can trigger both hapten and p-i responses.
If p-i HLA interaction is analogous to alloreactivity [30], then there is a potential to consider therapeutic options in SCAR based on clinical data of graft-versus-host disease (GVHD). So far, supportive care is the only treatment with good evidence for SJS/TEN and there is a lack of evidence for immunosuppressive agents. Given immunosuppression, in particular T-cell selective cyclosporine, is commonly used to prevent and treat GVHD, it will be important to clarify whether it also has a therapeutic role in SJS/TEN. While its efficacy is yet to be proven, data from small studies so far are encouraging and warrant further studies [4142].
Finally there may be role for rapid drug removal since p-i interaction is labile and reducing drug concentration can have potential clinical benefits. This possibility is also supported by a recent study that showed increased plasma concentration of oxypurinol is associated with poor outcome in allopurinol-induced SCAR [43]. Furthermore, Garcia-Doval et al. [44] have shown that the risk of death in SJS/TEN is associated with causative drugs with long half-life and with stopping the drugs late. These data point to the important role of drug concentrations in SCAR outcome. Since T-cell response is likely dose dependent in p-i concept as was shown for allopurinol specific T-cell response [45], studies on the benefit of rapid drug removal would be worthwhile.
ALTERED PEPTIDE REPERTOIRE
Altered peptide repertoire hypothesis proposes that a drug can interact with HLA class I molecules in a specific and noncovalent fashion and leads to presentation of altered peptides which elicit immune reactions. In doing so, there is an alteration to the stereochemistry of the antigen-binding cleft of the HLA molecule that modulates peptide repertoire, if the drug binding occurs in the endoplasmic reticulum (Fig. 1G) [28]. The presentation of altered self-peptides then leads to a T-cell response manifesting as a DHR. In contrast, p-i concept states that the formation of drug-pHLA complexes alone can activate T-cell immune response without requiring a specific novel peptide ligand. Altered peptide repertoire model shares some similarities with p-i concept in that a drug-pHLA complex plays an important role [29]. As discussed later, it can be regarded as a subset of p-i concept but with the main key difference being the binding of novel self peptides to drug-HLA complex.
There is little doubt that abacavir noncovalently binds to peptide binding groove of HLA-B*57:01 resulting in altered peptide repertoire as this has been proven via crystallography and peptide-elution studies [282946]. It is suggested abacavir is taken up by APC and enters its endoplasmic reticulum allowing noncovalent bonds within the bottom of the antigen-binding cleft, occupying binding pockets C, D and E, and protruding into the F-pocket. The binding is away from the site of peptide-TCR interaction. The resulting conformational change includes alteration in the shape and chemistry of the antigen-binding cleft. The repertoire of endogenous peptides that can bind HLA-B*57:01 undergo marked shift as a large proportion of the normal repertoire of peptides bound in the absence of abacavir would clash with abacavir moieties occupying the binding pockets. For example, the presence of the cyclopropyl group within the F pocket leads to a preference for a small aliphatic amino acid (valine, isoleucine, and leusine) and a peptide that conventionally binds a carboxy-terminal tryptophan at the anchor residue of HLA-B*57:01 is disfavoured [2829]. These novel peptides containing immunogenic neo-epitopes may be capable of inducing CD8+ T-cell response.
The subtle differences in contact residues that HLA-B*57:01 has with HLA-B*57:02, HLA-B*57:03 and HLA-B*58:01 make it a unique and highly specific for abacavir binding. Epidemiology studies made affirmative observations that African Americans who have higher frequencies of HLA-B*57:02, HLA-B*57:03, and HLA-B*58:01 are less affected by abacavir hypersensitivity syndrome in prevalence compared to those in Europe where HLA-B*57:01 is more common [47]. The positive predictive value (PPV) of HLA-B*57:01-positive patients treated with abacavir developing hypersensitivity is 47.9%, implicating there could be other factors involved in the development of DHR [48].
Self peptides bound to HLA-B*57:01 in the presence of abacavir stimulate abacavir-responsive T cells via polyclonal TCR usage [28]. As such, the normally self-tolerant T cells are exposed to novel neo-self peptides, mimicking allogenic antigen presentation to T cells [2530]. The immunogenic complex of novel self peptide-abacavir-HLA-B*57:01 was in effect a novel HLA-B allele presenting self-peptides to which the host has not been toleralised. The analogous process is thought to be the basis of graft-rejection and GVHD.
It was also hypothesised that abacavir-reactive T-cell response is due to the cross-reactivity of T cells primed from an earlier exposure to another foreign antigen [49]. However, it must be noted that abacavir specific T-cell responses are found in both memory and naïve T-cell populations, arguing against previous priming event as an absolute requirement [3049]. In light of naïve T-cell response to abacavir, allo-immune response may be a better suited model to explain DHR due to abacavir since T-cell allo-response does not necessarily rely on a previous priming event.
While HLA associated DHR has profound implication in individualised treatment to select out at risk patients to prevent DHR, the clinical relevance of altered peptide repertoire is yet to be proven as there are several criticism to this model. Firstly, it is not convincingly proven both in vivo and in vitro that altered peptide repertoire to be a general mechanism for small-molecule drugs binding to polymorphic HLA molecules causing DHR beyond the example of abacavir. Even in case of carbamazepine-induced SJS/TEN where a smaller magnitude of altered peptide repertoire occurs, T-cell response is shown to depend on p-i mechanism [2836]. Furthermore acyclovir can also induce a small shift in peptide repertoire in HLA-B*57:01 but cannot induce T-cell response [50]. Therefore it is possible that altered peptide repertoire may be a marker of drug binding to peptide binding groove rather than pathogenic per se. Secondly, Adam et al. [33] demonstrated that the addition of abacavir to drug reactive TCC elicited a rapid response in a percentage of TCC and these immediate reacting TCC also recognised abacavir pulsed APC. Since immediate reactivity can only be explained by p-i concept, altered peptide repertoire is not an explanation for reactivity to these TCC even though they are also stimulated by abacavir pulsed APC. Furthermore, it is currently unclear how altered peptide-abacavir-HLA-B*57:01 complex actually stimulates T cells; the unproven peptide specificity casts a shadow of uncertainty on the altered peptide repertoire model. In fact, some TCC that have delayed response to abacavir were shown to be poly-specific in that they also had allo-response to HLA-B*58:01 [30]. This implies that altered peptide specificity may be irrelevant since abacavir induced TCC are poly-specific and altered conformation of pHLA and not altered peptides are stimulatory. It is not so much which 'peptide' that TCR recognises but rather what conformation of pMHC complexes that TCR recognises.
ISSUES IN CHARACTERISING IMMUNE MECHANISMS
There are several issues in characterising the above described mechanisms of T-cell-mediated DHR. Firstly p-i mechanism and altered peptide repertoire are not mutually exclusive. Alteration in self peptide repertoire takes place intracellularly in endoplasmic reticulum and this is a peculiar phenomenon that was not anticipated from earlier studies of p-i concept as all other noncovalent interactions occur rapidly on the cell surface. Consequently altered peptide repertoire is delayed and processing-dependent, and washing of pulsed APC do not abrogate T-cell response [3351]. It is possible that T-cell response to altered peptide repertoire may be unique to abacavir and HLA-B*57:01 as it has not been described in other DHR. In addition, abacavir can directly bind to peptide-HLA-B*57:01 complexes on the cell surface in a manner that is typically described for p-i concept [33]. This phenomenon can be explained by p-i HLA model as described above (Figs. 1E, 2). Since the binding site and the mode of binding is identical for both of these mechanisms, one could argue that altered peptide repertoire can be classified as an 'atypical' subset of p-i concept. In this instance, alteration in peptide repertoire is simply due to the location of abacavir binding to HLA-B*57:01 rather than due to different mechanisms per se. It is intriguing that some TCC can recognise both abacavir-pulsed APC and abacavir in solution in the presence of HLA-B*57:01 [33]. This phenomenon is difficult to explain if we assume that altered peptide repertoire and p-i concept is mechanistically different since soluble abacavir cannot result in immediate alteration in peptide repertoire. On the other hand, if we assume altered peptide repertoire is a subset of p-i concept, then this observation can be readily explained since certain peptides may be present by HLA-B*57:01 both in the absence and presence of abacavir but with different configurations.
Some drugs such as penicillins and sulfamethoxazole can be presented by both hapten and p-i mechanism [93252]. By definition hapten and p-i mechanisms are mutually exclusive; one involves covalent bond and the other does not. However, these mechanisms can occur simultaneously in an individual and since they can both elicit T-cell responses, the clinical consequences may be indistinguishable. Since haptenation can occur on the cell surface to an existing pHLA complexes, processing-independent drug presentation does not disprove hapten mechanism [1953]. Secondly, drug-protein conjugate formation is a time dependent process and hence hapten mechanism is typically delayed [8]. However, p-i concept can also be delayed in some instances as was shown for lower avidity TCC to abacavir in HLA-B*57:01 [33]. Mass spectrometric analysis can prove that haptenation occurs but this does not necessarily mean that such haptens are responsible for a particular T-cell response. Indeed, haptenation to penicillins occurs in all individuals but only a minority will develop DHR. On the other hand, apart from altered peptide repertoire, there is no structural data that proves p-i concept actually occur. Nonetheless given abundant functional data, there is little doubt that p-i concept is real. Finally there are conflicting data on flucloxacillin-induced liver injury and HLA-B*57:01 as some TCC can recognise flucloxacillin via hapten mechanism and the others via p-i mechanism [93254]. It is likely that p-i mechanism is predominantly responsible for HLA-B*57:01 restricted response to flucloxacillin but which of these are functionally relevant is difficult to prove as mechanistic studies are only reliant on ex vivo or in vitro analysis [3255].
While there has been significant advances in understanding the mode of drug binding to TCR and/or HLA, why a same drug can cause different clinical manifestations in different individuals is unknown. For example, allopurinol is known to cause MPE, SJS/TEN and drug reaction with eosinophilia and systemic symptoms (DRESS) in different individuals yet only p-i mechanism is involved [27]. Furthermore cytokine analysis of in vitro stimulated peripheral blood mononuclear cells (PBMC) from drug-allergic donors shows variable patterns, making it difficult to associate certain cytokines with a clinical manifestation [56]. Granulysin is thought to play an important role in SJS/TEN and drug stimulated PBMC from SJS/TEN donors have been shown to secrete granulysin [3657]. However, a recent study by Chung et al. [58] seems to suggest that T cells cultured with oxypurinol in allopurinol induced DRESS patients also express granulysin implying that it may not be specific for SJS/TEN.
DANGER HYPOTHESIS
Danger hypothesis was first proposed by Matzinger [59] in the 1990s as an alternative to the self-nonself hypothesis. It asserts that immune responses are triggered by endogenous cellular alarm signals form distressed or injured cells. These danger or damage signals had been described by Matzinger [59] and expanded by subsequent investigators [60]. These are listed in Table 2. Many of these molecules were also found to bind to toll-like receptors, acting as an alternative to pathogen-associated molecular patterns for activation of APC and costimulation of T cells (signal 2) to initiate an immune response [61].
The danger hypothesis is not mutually exclusive with the above discussed concepts. The danger hypothesis can possibly explain why not all drugs that covalently bind to protein as in happen theory are associated with a significant incidence of DHR. In detail, only when a reactive metabolite causes cell damage and acts as a danger signal (signal 2), an immune response will be initiated. The signal 1 in this instance is provided by the TCR engagement with a drug modified peptide presented on MHC [62]. However if the signal 2 is missing, there is immune tolerance. This raises the question whether a concurrent event of other nature that causes cellular damage, for example a viral infection will act as a costimulation danger signal to induce DHR. In fact, the increased risk of DHR is observed in patients with mononucleosis taking ampicillin and human immunodeficiency virus (HIV) positive patients taking sulphonamides and other drugs [61]. Even physical injury in the form of surgery can increase procainamide-induced agranulocytosis [63].
The postulated participation of danger signal in costimulation can make DHR unreproducible and idiosyncratic as it is difficult to identify the exact danger signal and the source of the danger signal involved at the time of the initial reaction. In addition, it is difficult to replicate the danger signal in a controlled testing environment. This may contribute to the often inconsistent clinical observation of a well-documented genuine DHR to a culprit drug but negative skin testing and oral challenge to the same drug. Finally it must be emphasised that while danger hypothesis is highly plausible and is the likely explanation for why most patients do not develop DHR even when hapten is formed in all individuals, there are no convincing in vitro data for its pathogenic role.
The danger signal hypothesis may be relevant in certain DHR situations, but its necessity in inducing an immune reaction was recently questioned by the allo-immune model of DHR. As shown by Adam et al. [30], abacavir stimulation occurred in the absence of dendritic cells. The allo-immune stimulation provided by the abacavir-peptide-HLA B57:01 complex was stimulatory enough without need of 'danger' as cofactor. The massive direct T-cell stimulation by an allo-allele-like HLA peptide complex does not rely on prior antigen exposure nor on the signal 2, but only on direct TCR-allo-antigen contact [25]. If a drug indeed induces such an allo-like immune response, it would be a sufficient explanation for the overwhelming immune responses seen in DRESS or SJS/TEN without need of costimulation. Indeed the clinical picture of DRESS or SJS/TEN occurs also in the frame of GVHD.
RETHINKING THE DEFINITION OF TYPE B ADR
The widely accepted ADR classification into types A and B reactions was proposed almost 40 years ago by Rawlins and Thompson [64]. Due to limited understanding of immune mechanisms involved in DHR at the time, type B ADR was defined as 'bizarre' as it appeared unpredictable and dose-independent. However, with better understanding of DHR, type B ADR are becoming increasingly predictable. In the abacavir hypersensitivity syndrome, the PPV of HLA-B*57:01 is approximately 47.9% [48], whereas for allopurinol-induced SCAR, the PPV of HLA-B*58:01 is low at 2.7% in an unselected population, increasing to 18% in patients with chronic renal insufficiency [6566]. This suggests that identification of additional risk factors can increase predictability. In most cases of strongly associated HLA-associated DHR, negative predictive values of HLA tests are close to 100% [65]. This means some type B ADR can be 'predictably' ruled out by HLA typing. Screening for HLA-B*15:02, HLA-B*57:01 and HLA-B*58:01 testing prior to drug initiation can significantly reduce the risk of SCAR induced by carbamazepine, abacavir and allopurinol, respectively [486768]. Likewise it may be important to consider HLA allele frequency in drug research and development as clinical safety and tolerability in one population do not necessarily guarantee similar findings in other populations. Conversely, a drug which causes immune mediated DHR in one population may be better tolerated in another population especially if there is a strong HLA association with DHR.
The development of MPE in patients taking ampicillin during acute Epstein–Barr virus infection is also highly predictable as most exposed individuals develop the rash. While many of these patients can subsequently tolerate the drug once the acute infection has resolved, the fact that the drug is still required for the development of MPE suggests that it is a form of DHR, albeit transient. In addition, some patients subsequently develop persistent ampicillin allergy and the clinical manifestations are indistinguishable from MPE caused by other drugs which would be classified under type B ADR. Similarly trimethoprim-sulfamethoxazole hypersensitivity is estimated to occur in up to 60% or higher in HIV patients. This implies that certain infections can make DHR more predictable. Despite these strong associations, how infections interact with a drug and the immune system is yet to be clarified. Therefore one could argue that 'unpredictability' of type B ADR is mere reflection of the lack of understanding of the underlying process rather than intrinsic unpredictability of the drug to interact with the immune system.
Secondly type B ADR are thought to be dose independent. However, most in vitro data suggests that all immune mediated ADR are, in fact, dose dependent. Detailed piperacillin specific TCC analysis showed that T-cell reactivity was dependent on piperacillin dose [18]. Furthermore mass spectrometric analysis also confirmed that albumin modification with piperacillin was both time and dose dependent [8]. Since drug haptenation is dose-dependent, subsequently T-cell response is also dose dependent.
In case of p-i concept, dose dependent T-cell response was shown for oxypurinol, abacavir, carbamazepine and radiocontrast media [22333645]. This is not surprising since drug affinity to either TCR or pHLA is a determining factor in p-i concept and drug affinity is expressed by KD = [drug][receptor]/[complex]. Finally, Presentation of certain self-peptides on HLA-B*57:01 is enhanced by abacavir in a dose dependent manner [46]. Consequently, abacavir specific T-cell responses were also shown to be dose dependent [3351]. Putting it all together, the in vitro data suggest that DHR are highly dose dependent. Indeed, desensitisation procedure would be difficult to explain if DHR is a dose independent process.
As our understanding of immune mechanism in DHR improves, it is becoming increasingly clear that the current definition of type B ADR is inaccurate. It will become increasingly difficult to distinguish between types A and B reactions by relying upon clinical idiosyncrasy and dose-independence as the defining criteria. Therefore it will be important to rethink the definition of type B reactions and, more broadly, how we ought to classify ADR.
CONCLUSION
There have been significant discoveries and interest in recent years surrounding the immune mechanisms involved in T-cell-mediated DHR. Each of the above discussed mechanisms, namely hapten theory, p-i concept, altered peptide repertoire and danger hypothesis has its own strengths and limitations in explaining DHR. It is also important to note that some of these mechanisms are not necessarily mutually exclusive. They may even work synergistically in the process.
There are still many areas that await further clarification even with increasing understanding of the immune mechanisms. The other host and environmental factors that influence clinical phenotype and penetrance is still poorly understood. The clinical implication of DHR is profound as the associated conditions often carry significant morbidity and mortality and may represent models for more common diseases. Personalised medicine for prevention of DHR is now within the realm of evolving clinical practice with ongoing active research and development. In light of new gained insight, it would be timely to rethink the definitions and classifications of type B ADR as they are, indeed, becoming less 'bizarre'.



 XML Download
XML Download