

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Aeroallergens had been suggested as trigger factors for the development and exacerbation of asthma in 1860 [1]; consequently, the main immunologic mechanism of asthma was established as an axis of allergens, T helper 2 (Th2) lymphocytes, IgE, mast cells and eosinophils in the early 1990s [2]. Over half of adult asthmatics show negative skin prick reactivity to aeroallergens; therefore, the innate immune response emerged as a newly identified contributor to the immunopathogenesis of allergy and asthma in the mid-1990s [3]. Many inflammatory mediators are shown to be responsible for the maintenance and chronicity of inflammatory responses through the secretion of cytokines from epithelial, endothelial and constitutive mesenchymal cells in addition to immune cells [4]. Innate immune responses have also attracted significant attention as a candidate pathogenesis of asthma. Pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) are essential components of the innate immunity via activating several kinds of immune cells such as dendritic cells and activating inflammasome. However, the participation of inflammasome in the development and exacerbation of asthma has not been fully studied and remains controversial. We discuss the possible role of inflammasome in the development and exacerbation of asthma.
DAMP AND PAMP: INTERACTION WITH PATTERN RECOGNITION RECEPTORS
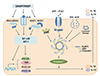
Pattern recognition receptors (PRRs) are expressed by innate immune cells that identify endogenous and exogenous PAMPs or DAMPs [4]. PRRs are divided into membrane-bound and cytoplasmic PRRs (Table 1). Membrane bound forms include Toll-like receptors (TLRs) and C-type lectin receptors. Nucleotide-binding oligomerization domains (NOD)-like receptors (NLRs) as well as RIG-I-like receptors (RLRs) located in the cytoplasm. Recognition of extracellular or endosomal PAMP is mediated by TLRs [5]. Lipopolysaccharide, cell components of gram negative bacteria, is one prototype of PAMP, and recognized by TLR4. Other PAMPs include bacterial flagellin (recognized by TLR5), lipoteichoic acid of gram positive bacteria, and single or double-stranded RNA and DNA of viruses (recognized by TLR3). Activation of TLRs induces nuclear factor (NF)-κB signaling to phosphorylate MAP kinase and the secretion of proinflammatory cytokines such as tumor necrosis factor (TNF)-α, IL-6, IL-8, pro-IL-1β, pro-IL-18 and costimulatory molecules (Fig. 1). Noninfectious DAMPs are released outside dyeing cells following tissue injury and amplify immune responses [6]. Protein DAMPs include heat-shock proteins, high-mobility group box1, and hyaluronan fragments. Nonprotein DAMPs include adenosine triphosphate (ATP), uric acid, heparin sulfate and DNA.
COMPONENTS OF INFLAMMASOME
Approximately 20 cytoplasmic NLRs and RLR respond to DAMPs and PAMPs. NOD1 and NOD2 recognize constituents of gram-negative or positive bacteria. There are 14 members of NACHT, LRR and PYD domains-containing protein (NALP). The activation process of NOD-like receptor family, pyrin domain containing 3 (NLRP3) is well known and its ligands are infectious molecules that include muramyl dipeptide, toxins, bacterial DNA, and double stranded RNA as well as noninfectious materials such as silica, alum, extracellular ATP and uric acid crystals. PAMPs are initially recognized by TLRs that activate NF-κB resulting in transcription of pro-IL-1β and pro-IL-18. Subsequently, the NLRP3 inflammasome complex is assembled to activate caspase-1, which cleaves pro-IL-1β and pro-IL-18 into their mature IL-1β and IL-18 (Fig. 1). In addition, caspase-1 activation induces interferon (IFN)-γ secretion [7] and cleavage of IL-33 [8].
EVIDENCES AND MECHANISMS OF INFLAMMASOME ACTIVATION IN ASTHMA
The administration of IL-1β induces airway hyperreactivity: a characteristic feature of asthma [9]. IL-1β is increased in the serum and bronchoalveolar lavage fluids of human asthmatics and decreases after steroid inhalation [10,11,12]. IL-18 levels are increased in theperipheral blood of asthmatics; however, controversial results have been reported [13,14,15]. The data suggest a possible presence of inflammasome activation in the asthmatic airways. Simpson et al. [16] recently proved the expression of NLRP3 and caspase-1 protein with a concomitant increase of IL-1β in macrophages as well as neutrophils in sputum of neutrophilic asthma.
Early exposure to microbes skews the immune response away from Th2 cytokines to prevent the development of IgE-dependent asthma [17]. The hygiene hypothesis raises the possible protective role of inflammasome against the development of asthma; however, current evidences favor a proinflammatory role of inflammasome activation. Components of virus, bacteria, and fungus are well known PAMPs. Double strand viral DNA activates the AIM2 inflammasome, while viral RNA such as Influenza A virus (IAV) triggers the assembly of the NLRP3 inflammasome [18]. Bronchial epithelial cells in asthmatics show an influenza-induced enhancement of caspase-1 activity [19]. In experimental mice, elderly ones have impairment of NLRP3 activity on IAV infection [20]. This data may extend to the high mortality of elderly subjects when they are infected with IAV. Similar to IAV, rhinovirus and respiratory syncytial virus trigger the NLRP4 inflammasome [21]. Chlamydia pneumonia, suspected as a causative agent for asthma chronicity, also activates the NLRP3/ASC (apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain) inflammasome [22].
Asthmatics have a different pattern of microbiome in the airways [23]. Interestingly, the lung microbial burden increases in asthma and chronic obstructive pulmonary disease and the extent of the total microbiomeis correlated to airway obstruction [24]. These data suggest that microbiome-induced inflammasome activation may contribute to asthma severity rather than the protective role of the hygiene hypothesis.
The critical role of inflammasome is highlighted in experimental asthma models rather than human asthmatics. During ovalbumin sensitization, alum potentiates the inflammatory response by activating the NLRP3 inflammasome [25]. The cytotoxic effect of alum adjuvant induces cell death and leakage of intracellular ATP or uric acid crystals and causes inflammasome activation [26]. Patients with asthma and chronic obstructive pulmonary disease have elevation of ATP as well as uric acid in their airways [27]; consequently, it is suggested that cell death occurs in the airways of symptomatic asthmatics or during prolonged exacerbation that may induce inflammasome activation.
Urban particulates and inorganic particles such as titanium dioxide, silica, and asbestos activate inflammasome [28]. PM10 or PM2.5 concentrations have been increasing, especially in developing countries and can contribute to increasing incidences and severity of asthma. However, the findings observed in animal models are yet to be validated in human asthmatic studies. Allen et al. [29] recently demonstrated that NLRP3 was not necessary in the development of allergic asthma models; however, they observed the effect of NLRP3 in Th2 type murine model of asthma. Subsequently, the Th1 or Th17 dominant asthma model is to be analyzed for the participation of inflammasome activation in different animal models of asthma.
IL-1β is synthesized by blood monocytes, tissue macrophages, and dendritic cell (DC) after activation by proinflammatory stimuli such as TNF or TLR ligands [30]. Inflammasomes are present in the myelomonocyte cell lineage; however, recent evidence indicates that several types of epithelial cells may also contain these structures [31]. IL-1β signaling regulates early Th17 cell differentiation via expressing interferon regulatory factor 4 and retinoic acid receptor-related orphan receptor gamma [32]; which is essential transcription factor in Th17 differentiation. IL-1β induces the development of IL-17-producing T cells in cooperation with IL-23 [33]. In contrast to IL-1β, IL-18 signaling affects IFN-γ producing Th1 cell differentiation in conjunction with IL-12 [34] and with IL-13 [35]. Among the multiple lymphocytes subsets, IL-1β and IL-18 in combination with the other cytokines may exert biologic effects on enhancement of Th17 cell differentiation in the lung leading to neutrophilic inflammation [36].
ASTHMA PHENOTYPES RELATED WITH INFLAMMASOME ACTIVATION
There are few studies on the relation of inflammasome activation with asthma phenotypes. Asthma is a heterogeneous group composed of different immunopathogenesis and clinical manifestations. Accordingly, a certain type of asthma may produce inflammasome activation as an underlying mechanism. Adult asthma can be classified into several subtypes: (1) early onset allergic asthma with specific IgE and Th2 cytokine dominance, (2) eosinophilic type of adult onset showing corticosteroid refractory, less allergic tendency but high IL-5 concentration, (3) exercise-induced mild intermittent type of Th2 dominancy and mast cell activation, (4) obesity - related adult onset without evidence of Th2 activation, and (5) neutrophilic types with low forced expiratory volume in 1 second and air trapping via Th17 pathway activation [37]. Adult onset asthma due to obesity may be related to the activation of inflammasome because inflammasome activation is observed in several metabolic disorders such as obesity [38]. Neutrophilic asthma is another group suspected of inflammasome activation. Airway neutrophilia is observed in noninfectious asthmatics, especially severe status asthmaticus [39]. Our previous studies indicate that refractory asthma is less allergic and had more intense airway neutrophilia compared to mild persistent asthma [40,41]. In the subgroup analysis of refractory asthma, airway neutrophilia is associated with severe air flow limitation [40]. The neutrophilic type (>70% of neutrophils in sputum) is more frequently observed in chronic progressive airway obstruction while eosinophilic type was predominantly observed in brittle types of asthma with frequent exacerbations rather than chronic progressive airway obstruction [41]. Moore et al. [42] recently reported that the multivariate approach identified 4 asthma sub-phenotypes that represent the severity and airway inflammation pattern. Among them, sputum neutrophil counts are associated with more severe asthma phenotypes. The extent of neutrophilic inflammation in asthmatic airways is closely correlated with gene and protein levels of IL-17A [43]. These data suggest that neutrophilic airway inflammation may be based on the axis of inflammasome activation-IL-1β/IL-18-IL-17-neutrophilic infiltration.
In addition, peripheral blood neutrophils express IL-17 protein and the levels are elevated in asthmatics [44]. In the analysis of sputum cells in neutrophilic asthma, NLRP3 and caspase-1 protein are expressed in neutrophils as well as macrophages [16]. Proteinase 3 from activated neutrophils during early responses directly cleaves pro-IL-1β and pro-IL-18 to IL-1β and IL-18 that contributes to the phenotype of subsequent adaptive immune responses independently from inflammasome activation [45]. These data suggest that neutrophils may exert an additional effect of initiating inflammasome activation and amplifying immune responses in asthmatic airways.
INFLAMMASOME-INDEPENDENT EFFECT OF DAMP AND PAMP ON IMMUNE RESPONSES
In allergic asthmatics, airway inflammation is triggered by the inhalation of specific allergens such as house dust mite allergens and pollens. DCs are essential for priming naive T cells towards Th2 differentiation; however, respiratory allergens also induce innate immune response via tight collaboration with PAMPs and DAMPs. Components of allergens trigger DCs in TLR4 or CTR dependent manner [46,47]. Without inflammasome activation, the triggering of these receptors on epithelial cells subsequently leads to the release of innate pro-Th2 cell cytokines that include thymic stromal lymphopoietin (TSLP), granulocyte-macrophage colony stimulating factor, IL-25, and IL-33 [48,49]. IL-33 represents a key cytokines in an innate immune response because it activates Th2 lymphocytes, eosinophils, basophils, and mast cells via up-regulation TSLP and IL-25 synthesis. Biologically active IL-33 is constitutively stored in the nuclei of human airway epithelial cells and endothelial cells and it can be released into the extracellular space after cellular damage [50,51]. Fungal allergen exposure induces an acute extracellular accumulation of ATP; autocrine ATP sustains increases in intracellular calcium concentration and releases IL-33 through the activation of P2 purinergic receptors [50]. Consequently, ATP and purinergic signaling in the respiratory epithelium are critical sensors for airway exposure to airborne allergens. IL-33 exerts its biological effect through similar signaling pathways that involve MyD88 and TRAF6 dependent pathways such as 1L-1β and IL-18, which are major end products of inflammasome activation. IL-33 stimulates Th2 cells through ST2 [52], a member of the IL-1 receptor family, which is expressed to various immune cells that include mast cells, basophils, eosinophils, Th2 lymphocytes, invariant natural killer T, natural killer cells, macrophages, dendritic cells, and neutrophils. IL-33 induces high amounts of Th2 cytokines by type 2 innate lymphoid cells (natural helper cells, nuocytes, or innate helper 2 cells) [53]. It was initially believed that processing of IL-33 by caspase-1 to a mature form was required for biological activity [54]. However, full-length IL-33 is biologically active and the processing of IL-33 by caspases results in its inactivation rather than activation [8,55].
Proteinase 3 from activated neutrophils is also noted to have dual functions on inactive human and mouse precursor IL-33 proteins [56]. Proteinase 3 activates the precursor form to biological active forms; however, the increase of incubation time with proteinase 3 abrogates IL-33 activities. The proteinase 3 directly cleaves pro-IL-1β and pro-IL-18 to IL-1β and IL-18 without inflammasome activation [45]. Considering together that neutrophils synthesize IL-17 [44], neutrophils may have potentials to initiate inflammasome activation and modulation of IL-33 leading to amplification of immune responses in asthmatic airways. Further researches are mandatory to solve the role of neutrophils in asthma, especially neutrophilic airway inflammation.
S100A9 IN NEUTROPHILIC ASTHMA
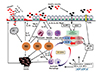
S100 protein family is a multigenic family of calcium modulated proteins that have a function of DAMP [57]. S100A9 belongs to the large Ca2-binding S100 protein family with the potential to form a complex with S100A8 (S100A8/A9: calprotectin), and exerts movement and activation of neutrophils [58]. It is one of the most abundant proteins found in neutrophils, macrophages, and epithelial cells during chronic inflammation. S100A8/A9 protein complex represents the exclusive arachidonic acid (AA)-binding proteins, which are important mediators of asthma, in human neutrophils [59]. AA derived from the complex could be used to generate other inflammatory mediators such as leukotriene B4, which trigger leukocyte activation and damage of vascular endothelium and smooth muscle cells, thereby exacerbating inflammation [60]. Recently, Kang et al. [61] found that S100A9 protein stimulates human bronchial epithelial cells to induce MUC5AC protein, which is the main mucin protein in asthmatic airways, via TLR4 activation. A previous proteomic study of ours [62] demonstrates increased levels of S100A9 in sputum of neutrophilic patients with uncontrolled severe asthma compared to those with controlled asthma (Fig. 2). Elevated S100A9 levels are limited to patients who are refractory to high doses of inhaled steroids; consequently, the function of S100A9 is suggested to relate with the resistance of airways to steroid treatment in severe asthma. In our study, S100A9 stimulates the activity of NF-κB and inflammasome activation (Fig. 3). IL-1β, a product of Inflammasome activation, also stimulates S100A9 induction by monocyte cell lines (Fig. 3). Thus, inflammasome stimulate inflammatory cells and epitheial cells to synthesize S100A9, which in turn could trigger activation of inflammasome by leukocyte leading to amplification of inflammation (Fig. 4).
Interestingly, S100A9 is revealed to promote proliferation of fibroblasts and up-regulate expression of collagen type III, α-smooth muscle actin and receptor for advanced glycation end-product [63]. Consequently, S100A9 may be a candidate that initiate and amplifies the neutrophilic inflammation of asthma as well as airway remodeling in collaboration with IL-17 (Fig. 4).
CONCLUSION
Neutrophilc inflammation of asthma has distinguished clinical features such as severe asthma and steroid resistance. This pattern may originate from the exaggerated activation of inflammasome, Th17 over-activation, and neutrophilic inflammation. In addition to caspase-1 activation, proteinase 3 and other protease from activated neutrophils directly cleave pro-IL-1β and pro-IL-18 to IL-1β and IL-18 that contribute to the phenotype of innate immune response. Neutrophilics in asthmatic airways may exert an additional effect of initiating inflammasome activation and amplifying immune responses. Among the several mediators from neutrophils, S100A9 seems to be one of candidate mediators to explain the action of neutrophils in amplifying neutrophilc inflammation. Further research in this area should focus on the potential for S100A9 in the induction of airway remodeling and steroid resistance, especially neutrophilic asthma.



 XML Download
XML Download