

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Why does drug eruption occur? Why does inflammation aggravate even if administration of the culprit drug is stopped? Unfortunately, we have no answers to these questions. However, the recent technical approach of the gene expression analysis has provided a great deal of information to solve them. Genome-wide analysis has revealed that individuals with certain human leukocyte antigen (HLA) polymorphisms have a 10 to more than 1,000-fold risk of drug hypersensitivity [1-4]. It will provide us with an order-made treatment to prevent severe drug hypersensitivity. Furthermore, recent in silico analysis makes it possible to reveal exquisite interactions between HLA molecules and drugs in the drug-antigen recognition of T cells [5], thereby providing us with a new perspective for drug discovery without hypersensitivity. In addition, recent studies using gene expression analysis have enabled the search for aggravation factors of severe drug hypersensitivity [6], which provides us with a novel therapeutic strategy. The latest innovative techniques are providing a new landscape to examine drug hypersensitivity.
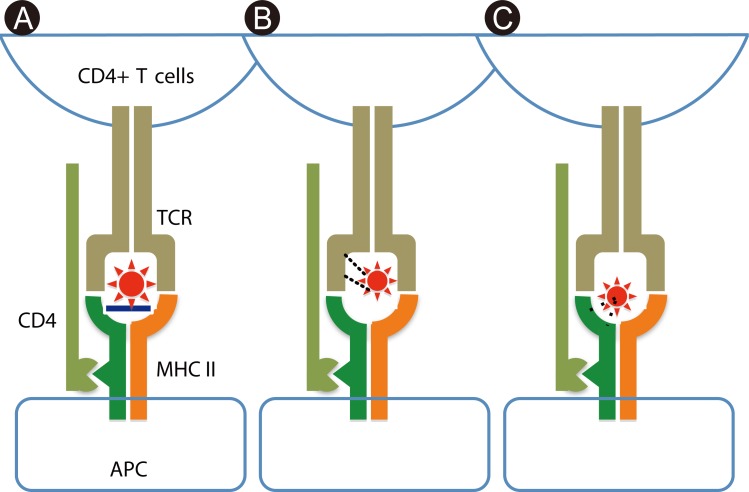
Mode of recognition of drug molecules in drug hypersensitivity
Because most drugs are small molecules, their covalent binding to self-derived proteins, such as membrane proteins and serum albumin, is necessary for immunogenicity. Essentially, a new epitope is presented by covalent binding of drugs with self-proteins, which is recognized by T cells as a haptenic antigen. It has been accepted as an important event for small molecules to be immunogenic to elicit contact dermatitis in experimental models [7], and may be central to the mechanism of drug allergies (Fig. 1A) [8]. However, it has been recently demonstrated that a high affinity of the drug for major histocompatibility complex (MHC) expressed on antigen-presenting cells (APCs), or for T cell receptors (TCRs) expressed on T cells, increases the possibility of activating T cells (Figs. 1B and C) [8]. Such binding is non-covalent with electrical or van der Waals forces, and can be easily dissociated by mechanical force. This type of interaction between T cells and APCs in drug recognition has been proposed as the pharmacological interaction concept (p-i concept) by Pichler et al. [9]. Interestingly, in several previous reports, anticonvulsant-specific T cell clones tend to have Vβ 5.1, despite the diversity of HLA alleles [10-13]. Under such conditions, antigen processing appears unnecessary for T cell activation because T cells activate even in the presence of APCs fixed with glutaraldehyde and the culprit drug [12]. Some clones usually do not require HLA matching of APCs for the response to the drug, indicating non-MHC restricted T cell recognition [14]. Such observations are accounted for by the "p-i concept" binding between the drug and TCR. On the other hand, in Han Chinese, individuals with HLA-B*1502 have a more than 2,500-fold risk of carbamazepine hypersensitivity [4]. Recent computer analyses of the molecular structure revealed an exquisite interaction between two molecules in the drug-antigen recognition. In such cases, the drug non-covalently binds to HLA-B 15:02, which is indicative of the "p-i concept" style binding [5].
In the hapten theory, after sensitization with the haptenized drug antigen, memory T cells recognize the antigenic epitope presented by APCs, with the arms of the TCR to critically fit its structure. However, the unique drug recognition manner in the p-i concept does not require the sensitization of T cells to the drug [9]. Basically, memory T cells that express TCRs with a certain affinity to the drug activate even if they are not sensitized to the drug, in the presence of the drug and APCs. Japanese individuals with HLA-A*3101 have a greater risk of carbamazepine hypersensitivity [3] in addition to Han Chinese with HLA-B*1502 [4]. Such cases show no correlation between this HLA polymorphism and the severity of drug hypersensitivity, suggesting that other factor(s) may contribute to exacerbate drug hypersensitivity. The size of the TCR repertoire of memory T cells is influenced by the individual's previous infections. The larger the size of the TCR repertoire of memory T cells, the higher the possibility of drug affinity for TCRs. Therefore, the number of T cells that bind a drug in the manner of the "p-i concept" may be increased in individuals who have experienced more infections. This scenario is well fitted with our clinical observations, although further investigation is needed to address this issue.
Characterization of drug-specific T cells
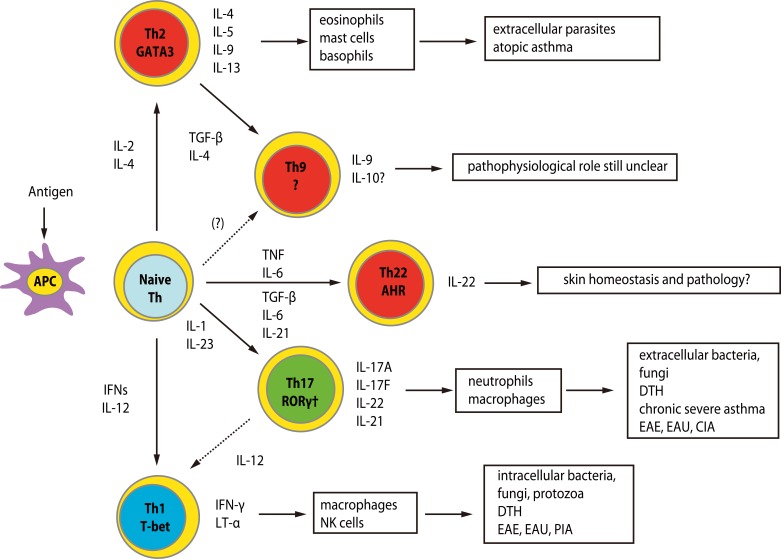
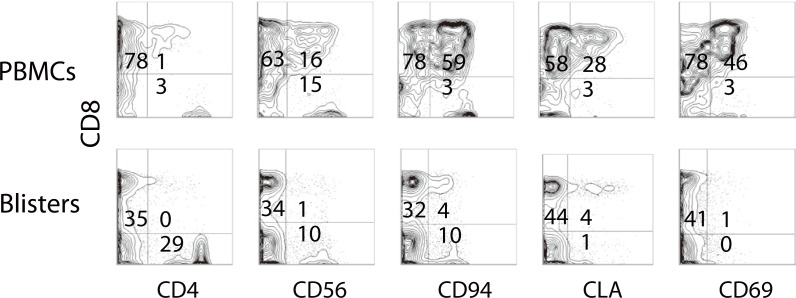
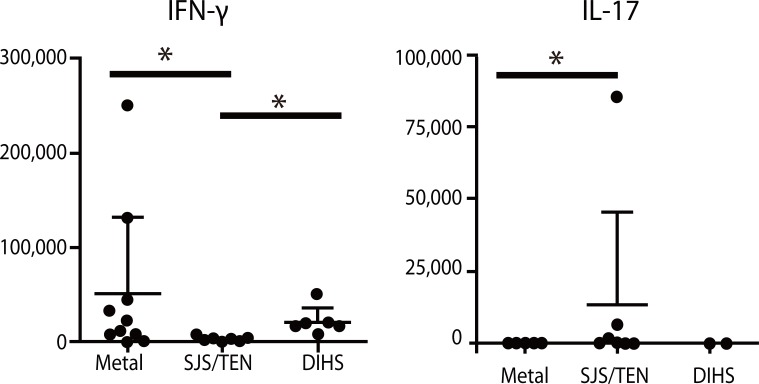
Effector T cells are divided into CD4+ and CD8+ subtypes, and the former is classified into Th0, Th1, Th2, Th9, Th17 and Th22, depending on the profile of released cytokines (Fig. 2) [15]. The quality and quantity of activated T cells reflects the clinical skin manifestations of drug hypersensitivity. The severity of CD8+ T cell infiltration into the skin is correlated with the degree of epidermal cell necrosis, which determines the clinical phenotype such as maculopapular eruption, Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN) (Fig. 3) [12]. In SJS/TEN, CD8+ T cells in blisters greatly activate to gain a natural killer (NK) cell marker, CD94/NKG2c, on their cell surface and acquire an antigen-independent killing activity resembling that of NK cells via binding to classical MHC (HLA-E) expressed on inflammatory epidermal cells [16]. Therefore, the activation status of CD8+ T cells in skin and blood is a predisposing factor for severe drug hypersensitivity such as SJS/TEN. On the other hand, a Th2-shifted immune response induces tissue eosinophilia and overproduction of IgE, resulting in the emergence of edematous erythema and wheals. β-lactam-specific T cells tend to produce Th2 cytokines such as interleukin (IL)-4, IL-5 and IL-13, which is indicative of Th2 cells and consistent with the clinical presentations [17]. IL-17-producing T cells, known as Th17 cells, are attracting attention as pathognomonic T cells in psoriasis, which also produce IL-8 (CXCL-8), a chemokine for neutrophils [18]. High serum levels of IL-17 and IL-8 are observed in acute generalized exanthematous pustulosis that clinically resembles pustular psoriasis, suggesting a contribution of Th17 to the pathology [19, 20]. Th17 cells cooperatively exaggerate the inflammatory responses of Th1/Tc1 [18]. Interestingly, CD4+ T cells with the ability to produce high levels of IL-17 are found among the drug-specific CD4+ T cell clones established from SJS/TEN patients (Fig. 4, unpublished data). Damaged epidermal cells in SJS/TEN release prostaglandin E2 and alarmins, which promotes differentiation and proliferation of Th17 cells activated by IL-23-producing dendritic cells after signaling via their receptors. This observation may be one of the explanations for the persistence of these serious diseases.
Regulatory T cells (Tregs) are another modifier of the inflammatory responses in drug hypersensitivity, which comprise natural and induced CD4+CD25+Foxp3+ Tregs and other cell types. Because of functional impairment of CD4+CD25+Foxp3+ Tregs [21-26], the risk of drug hypersensitivity would tend to rise in autoimmune disorders [27]. Anti-CCR4 antibody treatment is a novel therapy for adult T cell leukemia lymphoma (ATL/L) to kill CCR4-expressing ATL/L cells, while it may also reduce the number of CD4+CD25+Foxp+ Tregs that express CCR4. During such treatment, drug eruptions including SJS/TEN develop in around 70% of cases, suggesting that impairment of Tregs is a high risk factor for drug hypersensitivity. Functional impairment of Tregs has been found in drug-induced hypersensitivity syndrome (DIHS), which may be associated with a prolonged disease course [27].
Innate immunity and alarmins
Oppenheim proposed that damage associated molecular pattern molecules (DAMPs) released from damaged cells are cues for initiating immune responses in various organs by their activation after interacting with pattern recognition receptors and/or toll-like receptors [28, 29]. These molecules promote rapid recruitment of bone marrow-derived leukocytes to target tissues for inflammation and regeneration under various aseptic inflammatory conditions including SJS and TEN [6]. This molecular group contains various substances from simple chemicals, such as uric acid, to larger molecules such as IL-33 and DNA. Endogenous DAMPs are now designated as alarmins. High mobility group box-1 (HMGB-1), one of the most well-known alarmin members, is a non-histone protein with dual functions, namely transcriptional regulation by loose binding to chromatin inside cells and a cue for inflammation outside cells, with high potency to attract and activate various immunocompetent cells including monocytes and myeloid cells [28]. Recently, high expression levels of HMGB-1 have been found in blood from SJS patients [30]. Granulysin is a member of the saposin-like protein family, which is classified into 9- and 15-kDa isoforms. The 9-kDa granulysin is a well-described pore forming cytotoxic molecule with proinflammatory activity, and is considered as a crucial molecule to induce apoptosis of keratinocytes in SJS/TEN [31]. The 15-kDa granulysin has been recently accepted as an alarmin, which activates monocytes and dendritic cells via binding the toll-like receptor-4/Myd88, rather than a cytotoxic molecule [32, 33]. In addition, 15-kDa granulysin may act as an alarmin in SJS/TEN to promote Th17 cell responses by activation of dendritic cells.
Mechanisms of human herpes virus (HHV) reactivation in DIHS
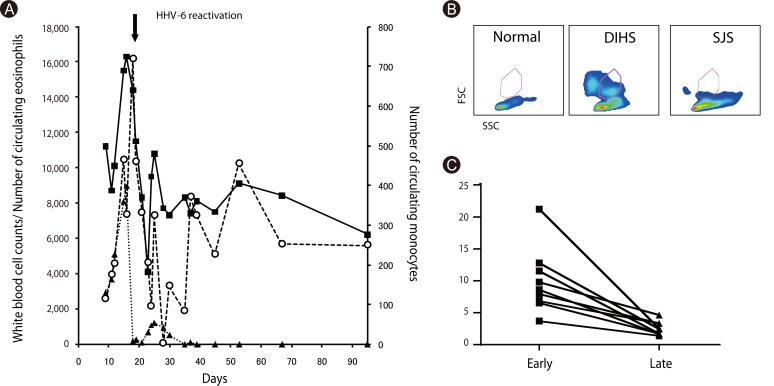
The most mysterious phenomenon in DIHS is reactivation of various HHV types, including cytomegalovirus, Epstein-Barr virus, HHV-6 and HHV-7, during the disease course [34, 35]. HHV-6 reactivation has been found in more than 60% of such cases in association with unfavorable outcomes [36]. Our concern is why HHV-6 frequently reactivates in DIHS. We found that the number of circulating monocytes transiently increase (>500/mm3) within 3 weeks and return to normal levels thereafter (Figs. 5A and C). The monocytes transiently observed in DIHS are different from conventional monocytes in terms of side scatter counts (SSC) and phenotype [37]. Monocytes from DIHS patients show higher SCC (Fig. 5B), suggesting enriched organelles, and comprise a minor CD14highCD16- population and a major CD14lowCD16+ population that express skin-associated molecules such as CCR4, CLA markedly and CCR10 partially (manuscript in preparation). This observation implies that they are a precursor mono/myeloid subset that differentiates into skin-resident macrophages. We also found the HHV-6 genome and virus structures in some of these monocytes (manuscript in preparation). Because some monomyeloid cells are latently infected with HHV-6 as virus reservoirs [38], monocytes that transiently circulate in DIHS patients appear to have originated from such cells. We found close contacts between monocytes and T cells in the skin, and the presence of HHV-6 in skin-resident CD4+ T cells (manuscript in preparation). HHV-6 infection of CD4+ T cells is an indispensable event for virus replication and reactivation [39]. Monocytes may lead this critical event in CD4+ T cells, suggesting that HHV-6 infects CD4+ T cells in the skin of DIHS patients. This notion provides new perspectives for understanding the pathology of DIHS to correlate allergy with viral infection.
Perspectives in future
The recent technical approach of gene expression analysis has provided a great deal of information to reveal the mechanisms of drug hypersensitivity. Consequently, a new approach with in silico analysis is being developed for clarification of the exquisite interaction between drugs and HLAs or TCRs. The Ministry of Health, Labour and Welfare in Japan recently reported that there were 131 deaths over 2.5 years owing to drug hypersensitivity. To overcome this iatrogenic death, we should make an effort to clarify the mechanisms and to establish an effective treatment. We believe that these advances may avoid drug hypersensitivity reactions in the near future.
 XML Download
XML Download