

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Vogt-Koyanagi-Harada (VKH) disease is a multisystem autoimmune disease that involves pigmented tissue in the eyes, auditory system, skin and central nervous system, without a history of penetrating trauma to the eyes. This disease mostly affected colored race, especially in Asia and America, frequently in women aged 20-50 years. It is rarely affected a child, but there was a reported case of a 4-year-old child with the disease. The exact cause of VKH disease is unknown, but there is a genetic predisposition. Patient with VKH disease usually presented with eye disorder which is a form of a bilateral granulomatous panuveitis. Ocular symptoms are often accompanied by extraocular organ involvement such as central nervous system, auditory system and integumentary system or skin. Extraocular organ involvement can be a headache due to cerebrospinal fluid pleocytosis, dysacousia, poliosis, and vitiligo [1-6]. VKH disease was first described by Vogt in 1906, in patients who experienced poliosis accompanied by bilateral uveitis, vitiligo, alopecia, and dysacousia. Furthermore, Koyanagi also reported a similar case in 1929, while Harada in 1926, described a patient with uveitis, exudative retinal detachment and associated with the presence of pleocytosis in cerebrospinal fluid [1].
CASE REPORT
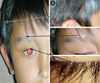
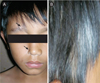
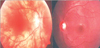
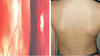
We report a boy 8 years and 9 months old, who visited Hasan Sadikin Hospital Bandung for a follow up visit, on April 1, 2010 with a chief complaint of blurred vision. The history taking obtained from the father informed us that the complaint began six weeks before examination without any previous eye trauma. It was accompanied with the patient's hair and eyebrows looked partly white, and appearance of white patches on the right forehead and right nasal. Previously, the patient had been treated in Cicendo, Eye Hospital Bandung, and then he was referred to our pediatric department for further treatment. Patient is currently treated with methylprednisolone. Two years before, his father noticed the patient right's eye was looked red when photographed, which was preceded by white patches on the right forehead. It then added with the hair, eyebrows and eyelashes become partly white and some white patches on the right nasal (Fig. 1). Patients came from families with low socioeconomic status. Physical examination revealed a good nutritional status, with vital signs was in the normal limit. There was vitiligo and atrophy in the right frontal and right nasal, poliosis on eyebrows, eyelashes, hair, and scoliosis (Figs. 1 and 2). Neurological examination showed a round anisocoric pupil diameter, 3mm on ocular dextra and 8 mm on ocular sinistra, with light reflex - / + and the impression of paresis on the 3rd, 4th, 6th, 9th cranial nerves (Figs. 2 and 3). Funduscopy examination, there was sunset glow appearance noted (Fig. 4). The visual acuity was right eye: 2/60, left eye: 1-0. Routine blood examination showed normal range with exception of elevated levels of total IgE: 1,570.4 IU/mL. Serology test performed with positive result for antitoxoplasma IgG, anti-Rubella IgG, anti-CMV IgG. Chest X-ray showed infiltrates suspected due to right pulmonary tuberculosis. Patient was diagnosed with Vogt-Koyanagi-Harada disease and then prescribed with methylprednisolone 1 mg/kg/day in three divided dose. This patient was also consulted to dermatology department and was prescribed with mometason furoate topical cream once daily. From the follow up control to ophthalmologist, the eye examination revealed an improvement on right eye panuveitis, with a remaining keratic precipitate in the endothelium, and minimal flare and cell on the anterior chamber. The patient still needed to continue his previous medication of homatropine eye drops three times daily and flumetholone eye drops every 4 h. ENT consultation revealed no ear inflammation or hearing disturbance.
DISCUSSION
We will discuss the problem of establishing the diagnosis, follow-up, procedure and prognosis of this patient. VKH disease occurs in this patient is a rare case, since most cases affect women in the third-fourth decade. In Indonesia, VKH disease has been known primarily among ophthalmologist, but not been widely known by other doctors. For that particular reason that this case report is intended for us as pediatrician to be able to recognize this disease in early stage and provide appropriate treatment so the patient will have a better prognosis.
The etiology of VKH disease is not certain, but several studies suspected an autoimmune role of T lymphocytes against melanocytes cells which triggered by certain factors. Prodormal symptoms indicate the possibility of herpes family infections (Epstein-Barr virus, Cytomegalovirus) as a triggering factor. Histopathological findings as well as in vitro experiments and animal models reinforce the importance of the CD4+ T lymphocytes in the disease pathogenesis. The main target of this disease is antigens derived from melanocytes which are tyrosinase and tyrosinase-related protein. Both enzymes are involved in the formation of melanin and are specifically located on melanocytes cells [1, 5]. VKH disease occurs in genetically susceptible individuals. This is apparently because some types of human leukocyte antigen (HLA) was reported to be associated with the reported incidence of this disease [7]. HLA-DRB1 * 0405 has a relationship with approximately 90% of patients with VKH disease in Asia and in other countries. From a study concluded that VKH disease was a disease with a combined allelic predisposition with HLA alleles DQA1 * 0301 as the primary and HLA-DRB1 * 0405 allele as an additional cause of this disease. Also found that HLA DQB1 * 0604 likely to have protective effects in this disease [7-10].
VKH disease has four clinical stages, prodormal, uveitic, convalescence, and chronic or recurrent . Extraocular manifestations usually occur in prodormal stage, convalecens stage, and chronic stage. Prodormal stage is characterized by presence of fever, nausea, and headache that lasts for 3-5 days. Nonspecific symptoms such as fever, and nausea also appear on the prodormal stadium. The auditory system involvement causing few symptoms such as dysacousia, tinnitus, high-frequency hearing disorders, and vertigo. While the involvement of the central nervous system, among others, would cause symptoms such as headache, meningismus, and pleocytosis that precede the ocular symptoms of 1-2 weeks. Although rare, focal neurological symptoms can occur such as paralysis of cranial nerves, and optic neuritis. Resemble symptoms of encephalitis can be found among others the existence of aphasia, personality disorders, decrease of consciousness, and seizure [1, 2, 6].
On uveitic stage, there will be the emergence of symptoms of photophobia, blurred vision and the presence of ocular pain. During examination we may find granulomatous bilateral anterior uveitis, with the precipitate of fat (mutton fat precipitates keratic) and nodules on the iris. We may also find choroiditis associated with the existence of exudative diffuse retinal detachment and hiperemic discus opticus. Cilliary body swelling can cause the diaphragm of lens-iris shifted anteriorly, causing narrowing of camera oculi anterior, and angle which results in an acute increase of intraocular pressure [1, 2, 6].
During convalescence stage, if patients receive proper treatment, eye symptoms and exudative diffuse retinal detachment will subside gradually. At this stage the fundus examination will reveal the form of 'sunset glow' (the fundus red-orange), with multiple depigmentation lesions of the retinal epithelium which scattered on the mid-peripheral fundus and a migration of retinal pigment epithelium. These indicate a severe aggression of melanocytes and pigmented tissue. On this stage we may find Sugiura sign, which is a depigmented limbus. Depigmentation can also be found on the skin such as vitiligo on the hands, shoulders, chest, back, face, poliosis (some part of hair, eyebrow, or eyelashes become white) and alopecia [1, 2, 6].
In the later stage, a chronic or recurrent uveitis may occur in 17-73% of patients. This recurrence usually presents in the form of anterior uveitis, but also reported in posterior chamber which signifies an ongoing aggression against the melanocytes. At this stage complications can occur in the eyes such as glaucoma, cataracts, choroidal neovascular membranes, and gliosis in the retina and choroid. In the event of complications, there will posible disturbance in the visual acuity [1, 6].
Uveitis in VKH disease occurs bilaterally, but in this patient we found only a chronic residual symptom of uveitis which only appears in the right eye. It is still possible due to damage to the cilliary body may not occur in both eyes so that the residual symptoms only appear in one eye. This was also reported by Kouda, 2002 and Usui, 2009 on their patients. These patients had unilateral uveitis which developed into bilateral uveitis, only three patients continued to show the character of unilateral VKH disease [11, 12].
This patient has reached the chronic stage of the disease because the patient has had disturbances of visual acuity accompanied with vitiligo and poliosis (Fig. 1). On further examination, we also found cranial nerve paralysis (Figs. 2 and 3) and the sunset glow on funduscopy examination (Fig. 4). It is presumed that two years prior when his father noted a red eye, the patients had symptoms of mydriasis in the right eye due to paralysis of the muscles in the cilliary body caused by previous uveitis. In this case, the patient was already in a chronic stage.
In 1980, the American Uveitis Society (Table 1) proposed diagnostic criteria by using the criteria proposed by Sugiura. However, these diagnostic criteria came short as it could not diagnose VKH disease in its early stage. So then, the International Committee on Nomenclature in 2001 proposed revisions to the diagnostic criteria for VKH disease, which divides patients into three categories: probable, incomplete and complete (Table 2) [1, 13-15].
VKH disease diagnosed based on clinical symptoms. In these patients the diagnosis is established using the criteria of the International Committee on Nomenclature who meet symptoms 1 - 3 and 4 + 5 so this is a complete VKH disease. Diagnosis of pulmonary tuberculosis in these patients also cannot be enforced, although tuberculin skin test and chest radiograph was performed, according to the tuberculosis scoring system only meet two criteria with a total score of 2. Anergy reactions in patients with autoimmune diseases is due to an increase in Th2 lymphocytes function and suppression of lymphocyte function of Th1 cells that play a role in the formation of a positive reaction on tuberculin skin test.
Suspicion of scoliosis (Fig. 5) excluded by tests carried out by colleagues from the orthopedic department. Orthopedic anthropometric measurements was within normal limits, therefore the asymmetry of the trunk is a normal variation arising from the habit of asymmetrical sitting.
Further investigations directed more to assess the existing eye disorders. We can perform ultrasonography, which will describe a reflective thickening of the choroid which is mild and diffuse, with the existence of a localized exudative retinal detachment. This distinguishes VKH disease with idiopathic effusion uvea and metastasis from carcinoma showing high reflectivity [1]. Investigation of fluorescein angiography on the acute phase will show choroiditis lession as a multiple hyperfluorescein dots that will unite to form an exudative retinal detachment. Shinzato, et al. [16] showed that the examination of anti Ro / SS-A showed a positive reaction in 10% of patients with VKH.
In this patient we found an increased total IgE levels, with differential count did not reveals any eosinophilia. This can lead to the existence of a hyper-IgE syndrome, but there is not enough data to support hyper-IgE syndrome diagnosis which one of them achieve total IgE levels above 2,000 IU/mL. So in this case, we should monitor the total IgE levels and clinical signs towards hyper-IgE syndrome [17, 18]. The serology test for CMV, Toxoplasma, Herpes and Rubella was conducted to find the trigger factors of VKH disease, and as a differential diagnosis the cause of uveitis. The results revealed the patient had the possibility of prior Rubella, CMV and Toxoplasma infection. Standard treatment for VKH disease is high dose systemic corticosteroids, and topical corticosteroids to treat anterior uveitis. Predisone can be used in an appropriate dose of 1-2 mg/kg/day peroral, with starting dose is 1 to 1.2 mg/kg/day. There is no evidence that indicates intravenous delivery is better than the peroral administration. Some centers are using of intravenous methylprednisolone. Steroid is given for 6-9 months prior to tappering off. This will give a better prognosis, decrease the duration of illness, decrease the incidence of convalescents stage, and decrease the extraocular manifestations [1]. Tappering off done by lowering the dose of prednisone 10 mg/day weekly until the dose reached 40 mg/day, followed by reduced 5 mg/day dose weekly to 20 mg/day, then 2.5 mg/day weekly until the dose reached 10 mg/day. Next tappering off done by lowering the dose of 2.5 mg/day per month. Therapy was performed at least as long as six months [13, 19]. Corticosteroids administration can be done in a longer time in a state of chronic uveitis in the anterior segment, up to 48 months [20].
In refractory cases, steroid administration should be discontinued immediately and replace it with other immunosuppressive drugs, such as cyclosporin (the most widely used after a steroid), cyclophosphamide, chlorambucil, azathioprine and mycophenolate mofetil. Cyclosporin dose is 3-5 mg/kg/day, whereas chlorambucil 0.1 mg/kg/day. We also must consider the side effects of these drugs which include the decrease amount of leukocytes, impaired kidney function and liver function [13]. Choroidal neovascular membranes can be treated with traditional photocoagulation or by using anti-angiogenic drugs. Closed angle glaucoma in the acute phase of VKH disease would improve after steroid administration steroid [1].
In this patients steroid treatment was given 1 mg/kg/day in three divided dose equal to 24 mg of methylprednisolone per day (methylprednisolone tablets 3 × 8 mg). We had to evaluate this therapy every two weeks to see any recurrence. The patient also needed evaluation from ophthalmologist and dermatologist. For further diagnosis, we planned for the patient to be consulted with ENT specialist to find whether there is a hearing disorder.
With administration of steroid drugs, two thirds of patients becomes able to maintain good visual acuity, with at least have a visual acuity of 20/40 or better, only a minority of patients (11%) having poor visual acuity (20/200 or better visual acuity). Treatment with corticosteroids can also control symptoms in almost all patients with uveitis. Patients will have a better prognosis if they have a good visual acuity at 1 month after onset, younger age at onset of disease, early treatment with corticosteroids [13, 19].
With early diagnosis and appropriate treatment, this disease is not expected to develop into chronic or recurrence stage. No mortality associated with VKH disease has been reported, thus the life prognosis is good. With the current treatment, patients expected to have a better prognosis, especially the visual acuity. But unfortunately this patient came in an advanced stage, with already decrease in the visual acuity. Thus, the functional prognosis in this patient was bad.



 XML Download
XML Download