

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
NADPH oxidase (NOX) is a membrane-bound enzyme complex that catalyzes the electron transfer from NADPH to molecular oxygen, as shown in the following reaction. NOX is a genuine reactive oxygen species (ROS) generator in the sense that ROS generation is the primary and sole function of NOX, unlike other sources of ROS, such as xanthine oxidase or cytochrome P450, which generate ROS as a byproduct of their reactions or after conversion to a dysfunctional state.1
NOX was originally found in phagocytic leukocytes, such as neutrophils and macrophages. It participates in respiratory burst during immune responses by producing superoxide. NOX in phagocytes is composed of membrane-bound catalytic subunits, such as gp91phox and p22phox, and regulatory cytosolic subunits, including p47phox, p67phox, p40phox, and Rac. NOX is latent under normal circumstances, but upon activation, its regulatory cytosolic subunits translocate to the membrane and associate with membrane-bound catalytic subunits. The assembled enzyme complex then produces superoxide via the one-electron reduction of the oxygen molecule by gp91phox using NADPH as the electron donor.2 During the last two decades, NOX activity has been reported in non-phagocytic cells and additional isoforms of gp91phox have been discovered. To date, seven NOX isoforms have been identified in humans and named NOX1, NOX2, NOX3, NOX4, and NOX5 and dual oxidase (DUOX) 1 and DUOX2. The originally identified phagocytic NOX has now been named NOX2. NOX exhibits tissue-specific expression patterns and distinct subcellular localizations that are dependent on isoforms. Furthermore, each isoform works with different regulatory subunits and has a specific regulatory mechanism; thus the isoforms have quite different pathophysiological functions.3
As its etiological roles have been uncovered, NOX has come to be viewed as a therapeutic target. Among NOX isoforms, NOX2 has been proved to be a therapeutic target for inflammation, diabetic complications, cardiovascular disorders, and neurodegenerative diseases, such as Alzheimer's and Parkinson's diseases.4 Accordingly, considerable effort has been put into developing NOX2 inhibitors. Not a few chemicals have been reported to inhibit NOX2. However, most have limitations in specificity and in vivo or clinical applicability, including non-specific ROS-scavenging activity, off-target effects, and poor pharmacokinetic properties.5 Consequently, they have yet to reach the clinic and improved NOX2 inhibitors are currently under development.
Schisandra chinensis (Turcz.) Baill. (Schisandraceae) is a deciduous woody vine native to Far East Asia. Its berries have been used in traditional oriental medicine. Indeed, various recent studies have revealed their diverse biological effects on the central nervous, immune, endocrine, and cardiovascular systems.6 In particular, the fruits of S. chinensis have anti-oxidant and anti-inflammatory activities in in vitro and in vivo studies. In addition, its active constituents, such as gomisin A and schisandrin B, are capable of inhibiting lipopolysaccharide-induced NOX activation and ROS production.78 Based on these previous reports, the NOX2-inhibitory effect was explored with the fruits of S. chinensis.
Experimental
General experimental procedure
Nuclear magnetic resonance (NMR) spectra were determined on a Varian 400 (Varian, Palo Alto, CA, USA) - 400MHz spectrometer at 400 MHz for 1H-NMR and at 100MHz for 13C-NMR. High-resolution mass spectra data were obtained from a Waters Xevo G2 Q-TOF mass spectrometer (Waters, Milford, MA, USA). Semi-preparative high performance liquid chromatography (HPLC) was performed on a Gilson 321 pump and Gilson 172 Diode Array Detector (Gilson, Middleton, WI, USA). The HPLC columns used were YMC-pack Ph, 250 × 20 mm (YMC, Kyoto, Japan) and Luna 5 µm C18 (20) 100A column 250 × 10 nm (Phenomenex, Seoul, Korea). Water was purified using a Milli-Q system (MilliporeSigma, Burlington, MA, USA). Column chromatography was performed on C-18 RP silica gel (Cosmosil, Kyoto, Japan) and Sephadex LH-20 (GE Healthcare, Uppsala, Sweden). TLC analysis was conducted on silica gel 60 F254 plates (Merck, Darmstadt, Germany). The spots were visualized by spraying with 10% aqueous H2SO4.
Plant materials
the fruits of S. chinensis was obtained from an oriental market, and the raw material (CYWDU-KP0009) used in the present study was deposited at the College of Pharmacy, Dongguk University-Seoul, Republic of Korea.
Extraction and Isolation
the dried fruit (5.8 kg) was kept at room temperature, extracted by MeOH three times, and evaporated in vacuo. MeOH extract (1.53 kg) was suspended in H2O and partitioned with CHCl3, EtOAc, and n-butanol successively to give the residue of 180 g of CHCl3-soluble extract, 132 g of EtOAc-soluble extract, 434 g of butanol-soluble extract, and 759 g of water-soluble extract. The portion of CHCl3-soluble extract (35 g, SCC) was chromatographed with silica column chromatography (5 × 90 cm, 600 g) using the gradient of increasing polarity with hexane-EtOAc (30:1–2:1) and CHCl3-MeOH (20:1–1:1) as solvents. The resultant eluent was fractionated into 17 sub-fractions (SCC-1-SCC-17). The SCC-3 fraction (42.4 mg) was chromatographed with Sephadex LH-20 column chromatography (2 × 90 cm) using the gradient with CHCl3-MeOH (1:1) as solvents and fractionated into three subfractions (SCC-3A and SCC-3C). The SCC-3C fraction was subjected to HPLC separation with MeCN-H2O (80:20) at 3.0 ml/min by isocratic elution for 20 min and then 100% MeCN for 6 min to afford compound 15 (tR 19.55 min, 17.2 mg). The SCC-5 fraction (162.3 mg) was subjected to HPLC separation with MeCN-H2O (70:30) at 3.0 ml/min by isocratic elution for 34 min and then 100% MeCN for 6 min to afford compounds 28 (tR 20.25 min, 4.2 mg), 16 (tR 30.25 min, 6.5 mg), and 17 (tR 33.10 min, 21.2 mg). The SCC-7 fraction (112.2 mg) was subjected to HPLC separation with MeCN-H2O (80:20) at 7.0 ml/min by isocratic elution for 35 min and then 100% MeCN for 6 min to afford compounds 18 (tR 21.0 min, 5.9 mg), 23 (tR 28.25 min, 1.2 mg), and 24 (tR 31.25 min, 27.1 mg). The SCC-8 fraction (71.1 mg) was purified by MPLC equipped with a reversed-phase (RP) column using a gradient mixture of MeOH-H2O (30:70–90:10), giving eight sub-fractions (SCC-8A-SCC-8H). The SCC-8H fraction (14.8 mg) was subjected to HPLC separation with MeCN-H2O (80:20) at 2.0 ml/min by isocratic elution for 20 min and then 100% MeCN for 6 min to afford compound 19 (tR 17.75 min, 2.1 mg). The SCC-9 fraction (576.4 mg) was purified by MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (5:95–90:10), giving 12 sub-fractions (SCC-9A-SCC-9L). Compounds 27 (6.5 mg) and 20 (105.6 mg) were obtained from SCC-9B and SCC-9F, respectively. The SCC-9G fraction (148.4 mg) was chromatographed with silica column chromatography (2 × 35 cm, 75 g) using the gradient with hexane-EtOAc (50:1–10:1) as solvents and fractionated into five sub-fractions (SCC-9G1-SCC-9G5). The SCC-9G3 fraction (24.3 mg) was chromatographed with silica column chromatography (1 × 10 cm, 7 g) using the gradient with hexane-EtOAc (10:1–2:1) as solvents and fractionated into six sub-fractions (SCC-9G3A-SCC-9G3F). Compound 21 (9.5 mg) was obtained from the precipitate from the SCC-9G3A fraction. SCC-10 (1.12 g) was chromatographed with silica column chromatography (1.5 × 45 cm, 90 g) using the gradient of increasing polarity with hexane-EtOAc (10:1–3:1) and CHCl3-MeOH (100:0–10:1) as solvents and fractionated into seven sub-fractions (SCC-10A-SCC-10G). The SCC-10B fraction (15.2 mg) was subjected to HPLC separation with MeCN-H2O (80:20) at 2.0 ml/min by isocratic elution for 11 min and then 100% MeCN for 6 min to afford compound 7 (tR 10.25 min, 2.0 mg). The SCC-10D fraction (305.5 mg) was purified by MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (40:60–90:10), giving 11 sub-fractions (SCC-10D1-SCC-10D11). Compound 2 (25.0 mg) was obtained from SCC-10D10. The SCC-10D6 fraction (20.1 mg) was subjected to HPLC separation with MeCN-H2O (70:30) at 7.0 ml/min by isocratic elution for 20 min and then 100% MeCN for 6 min to afford compound 1 (tR 18.25 min, 3.3 mg). The SCC-11 fraction (10960.3 mg) was purified by an MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (5:90–90:10), giving 20 sub-fractions (SCC-11A-SCC-11T). Compound 25 (5141.1 mg) was obtained from SCC-11A. The SCC-11H to SCC-11K fractions (241.3 mg) were combined and subjected to HPLC separation with MeCN-H2O (60:40) at 3.0 ml/min by isocratic elution for 30 min and then 100% MeCN for 6 min and afforded compounds 29 (tR 12.75 min, 5.5 mg), 11 (tR 17.25 min, 2.6 mg), 3 (tR 18.75 min, 7.7 mg), and 4 (tR 20.10 min, 9.7 mg). The SCC-12 fraction (2062.5 mg) was purified by an MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (5:90–90:10), giving 14 sub-fractions (SCC-12A-SCC-12N). The SCC-12J fraction (69.9 mg) was subjected to HPLC separation with MeCN-H2O (50:50) at 3.0 ml/min by isocratic elution for 34 min and then 100% MeCN for 6 min and afforded compound 5 (tR 34.75 min, 6.4 mg). The SCC-12K fraction (56.5 mg) was purified by MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (40:60–80:20), giving five sub-fractions (SCC-12K1-SCC-12K5). The SCC-12K4 fraction (36.3 mg) was subjected to HPLC separation with MeCN-H2O (60:40) at 3.0 ml/min by isocratic elution for 20 min and then 100% MeCN for 6 min and afforded compounds 8 (tR 10.55 min, 1.5 mg), 22 (tR 11.45 min, 2.7 mg), 9 (tR 14.25 min, 1.8 mg), and 10 (tR 16.45 min, 12.5 mg). The SCC-13 fraction (1495.6 mg) was purified by MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (5:90–90:10), giving 26 sub-fractions (SCC-13A-SCC-13Z). The SCC-13K to SCC-13M fractions (163.2 mg) were combined and then subjected to HPLC separation with MeCN-H2O (50:50) at 7.0 ml/min by isocratic elution for 16 min and then 100% MeCN for 6 min and afforded compounds 6 (tR 13.50 min, 15.8 mg), 12 (tR 15.75 min, 5.4 mg), and 13 (tR 17.55 min, 2.8 mg). SCC-13T was chromatographed with Sephadex LH-20 column chromatography (2 × 90 cm) using the gradient with hexane-EtOAc 100% MeOH as solvents and fractionated into two sub-fractions (SCC-13T1 and SCC-13T2). Compound 14 (32.0 mg) was obtained from the SCC-13T1 fraction. The SCC-15 fraction (1441.9 mg) was purified by MPLC equipped with an RP column using a gradient mixture of MeOH-H2O (10:90-90:10), giving 19 subfractions (SCC-15A-SCC-15S). Compound 26 (18.2 mg) was obtained from SCC-15B.
Dimethyl-malate (25)
Amorphous oil, 1H-NMR (CDCl3, 400 MHz): δ 4.53 (1H, t, J = 6.0, 4.4 Hz, H-2), 3.82 (3H, s, 1-OCH3), 3.73 (3H, s, 4-OCH3), 3.39 (1H, brs, 2-OH), 2.87 (1H, dd, J = 16.5, 6.2 Hz, H-3b), 2.80 (1H, dd, J = 16.5, 6.2 Hz, H-3a). 13C NMR (CDCl3, 100 MHz): δ 173.7 (C-1), 171.0 (C-4), 67.2 (C-2), 52.8 (4-OCH3), 52.0 (1-OCH3), 38.4 (C-20).
NOX2 activity assay
NOX2 activity was assessed in PLB-985 cells using an 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red; Molecular Probes, Eugene, OR, USA) fluorescence ROS indicator. PLB-985 cells were kindly provided by Dr. M. Dinauer (Washington University, St. Louis, MO, USA). Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin, and were maintained in a humidified chamber at 37 ℃ with a 5% CO2 environment. For assay, cells were washed and resuspended with Hanks' balanced salt solution (HBSS; 130 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2; pH 7.4) and 1 × 105 cells were aliquoted into a well of 96-well plate. Cells were pretreated with testing material for 60 min and NOX2 was stimulated with 200 ng/ml of phorbol 12-myristate 13-acetate (PMA; Santa Cruz Biotechnology, Dallas, TX, USA). ROS generation was detected with 10 µM of Amplex Red combined with 0.1 U/ml of horseradish peroxidase (HRP; Sigma-Aldrich, St. Louis, MO, USA). The fluorescence intensity was measured at 560/585-nm excitation/emission wavelengths for 60 min with 5-min interval using the SpectraMax M3 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Increase in fluorescence intensity was confirmed to be linear for more than 60 min.
Superoxide scavenging assay
Superoxide scavenging activity was tested in cell-free, xanthine/xanthine oxidase superoxide generating system. Superoxide was produced by 200 µM of xanthine and 0.01 U/ml of xanthine oxidase in HBSS with or without testing compound. Superoxide was quantified with 500 µM 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-1; Seoul Clinical Genomics, Seoul, Korea) by measuring the absorbance at 450 nm for 5 min using the SpectraMax M3 microplate reader (Molecular Devices).
HRP activity assay
Peroxidase activity was determined by a colorimetric method. The reaction mixture was composed of 0.003% H2O2, 0.5% o-dianisidine, and testing compound in 10 mM sodium phosphate buffer (pH 6.0). The reaction was initiated by the addition of 10 µl of 0.1 U/ml HRP to 90 µl of reaction mixture. The absorbance at 460 nm was measured at 15-s intervals for 5 min with the SpectraMax M3 microplate reader (Molecular Devices). Boiled HRP was prepared by heatinactivation at 99 ℃ for 15 min and used as a negative control. Relative HRP activity was calculated from the slope of the graph of increasing absorbance.
Statistical analyses
Means and standard error of means were calculated for all experimental groups. Data were subjected to one-way analysis of variance followed by Dunn's test to determinewhether the differences relative to the controls were significant. Statistical analysis was performed with SigmaPlot software ver. 13 (Systat Software, San Jose, CA, USA). P values of < 0.05 were considered statistically significant.
Result and Discussion
The NOX2-inhibitory effect was tested with crude methanolic extract and its organic solvent partitions, including chloroform-soluble, ethyl acetate-soluble, butanolsoluble, and water-soluble extracts. PLB-985 is a human diploid myeloid leukemia cell line that expresses NOX2 exclusively,910 which was also confirmed in this study by a real-time polymerase chain reaction (data not shown). PLB-985 was pretreated with 100 µg/ml of each extract for 60 min, and NOX2 was stimulated with phorbol 12-myristate 13-acetate (PMA). Among five extracts, the chloroform-soluble partition exhibited the highest inhibitory potential, which was followed by the ethyl acetate partition and butanolic partition, whereas the aqueous partition and methanolic extract showed minimal activity (Fig. 1A). The acknowledged NOX inhibitors diphenyleneiodonium (DPI) or VAS2870 were confirmed to work in this assay system at 10 mM (Fig. 1A, gray bars).
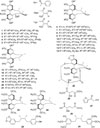
To determine the active constituents in the chloroformsoluble partition, repeated chromatographic separations were conducted. The twenty nine compounds were isolated and identified (Fig. 2). All structures of 1 – 29 were identified by the comparison of their spectroscopic data with those of the literature as the following: gomisin G (1),11 angeloylgomisin Q (2),12 tigloylgomisin P (3),13 schisantherin A (4),14 benzoylgomisin Q (5),15 (+)-gomisin T (6),16 schisandrol B (7),17 schisandrol A (8),1618 tigloylgomisin H (9),19 angeloylgomisin H (10),19 schinlignan D (11),20 (+)-gomisin H (12),21 schinlignan F (13),22 benzoylgomisin H (14),19 schisandrin C (15),2223 g-schizandrin (16),24 schisandrin B (17),23 gomisin L2 (18),25 (−)-gomisin L1 (19),23 gomisin J (20),23 (−)-gomisin K1 (21),26 gomisin D (22),12 dehydroschisandro A (23),27 schisandrin A (24),23 dimethyl-malate (25),28 2-hydroxy-5-methyl ester (26),29 2-(2-hydroxyacetyl)furan (27),30 anwulignan (28),31 and pregomisin (29)32 (Fig. 2).
These twenty nine compounds were screened for their NOX2-inhibitory activity. PLB-985 cells were pretreated with 10 µM of testing compounds, and PMA-stimulated ROS generation was detected with Amplex Red. Of these, lignan-type structured compounds 6, and 29 were identified as active inhibitors, which exhibited more than 40% inhibition (Fig. 1B). To calculate the half-maximal inhibitory concentration (IC50), the concentration dependency for NOX2 inhibition was tested and plotted (Fig. 3). The IC50 for compound 6 was approximately 9.4 ± 3.6 µM, and that for compound 29 was approximately 62.9 ± 11.3 µM, respectively.
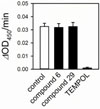
In addition to NOX2 inhibition, the nonspecific ROS-scavenging effect may lead to a false positive result in NOX2 activity assay. Hence, ROS-scavenging activity was tested in a xanthine-xanthine oxidase superoxidegenerating system. Superoxide produced by xanthine and xanthine oxidase was quantified with WST-1 in the presence or absence of compound 6 or 29. However, neither compound 6 nor 29 showed superoxide-scavenging activity at 50 µM, while the superoxide dismutase mimetic TEMPOL abolished superoxide nearly completely (Fig. 4). Therefore, compounds 6 and 29 are not nonspecific superoxide scavengers, but NOX2 inhibitors.
Since Amplex Red requires peroxidase to react ROS and to produce the red-fluorescent oxidation product, HRP was used in this assay. Therefore, the effect of compounds 6 and 29 on HRP activity was examined to test whether NOX2 inhibition observed in Fig. 1 and Fig. 3 is ascribed to artifact caused by HRP inhibition rather than NOX2 inhibition. Compound 6 inhibited HRP marginally; its inhibitory activity was minimal at 10 µM, although the extent of inhibition was approximately 39% at 100 µM (Fig. 5, left panel). Considering less than 10 µM of IC50, the attenuation of ROS generation shown in Fig. 1 and Fig. 3 should be mainly attributed to NOX2 inhibition. However, compound 29 was capable of inhibiting HRP substantially (Fig. 5, right panel). Although it is unlikely that HRP inhibition is directly proportional to NOX2 inhibition, HRP inhibitory activity may be involved in NOX2 inhibition observed in Fig. 1 and Fig. 3. According to these results, the efficacy on NOX2 might be lower than those calculated based on the data from Fig. 3, especially for compound 29. Further study will be needed to characterize the non-specific effect of compounds 6 and 29.
In conclusion, potential inhibitors for NOX2 were found from the fruit extract of S. chinensis. One of them, gomisin T (6), exhibited the highest efficacy with an IC50 of less than 10 µM. NOX2 inhibitory activity was also observed with pregomisin. Although several lines of chemical structure have been reported to inhibit NOX,4 lignan-type structures (6, 29) are proposed as NOX2 inhibitors for the first time. Gomisin T (6) and other constituents of S. chinensis were identified as promising NOX2 inhibitors. Further characteristics regarding structure-activity relationships, isoform specificity, the molecular mechanism of action, pharmacokinetic properties, and in vivo efficacy will be investigated in future studies.



 XML Download
XML Download