

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
This study investigated the effects of (−)-sesamin on memory deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned mouse model of Parkinson's disease (PD). MPTP lesion (30 mg/kg/day, 5 days) in mice showed memory deficits including habit learning memory and spatial memory. However, treatment with (−)-sesamin (25 and 50 mg/kg) for 21 days ameliorated memory deficits in MPTP-lesioned mouse model of PD: (−)-sesamin at both doses improved decreases in the retention latency time of the passive avoidance test and the levels of dopamine, norepinephrine, 3,4-dihydroxyphenylacetic acid, and homovanillic acid, improved the decreased transfer latency time of the elevated plus-maze test, reduced the increased expression of N-methyl-D-aspartate (NMDA) receptor, and increased the reduced phosphorylation of extracellular signal-regulated kinase (ERK1/2) and cyclic AMP-response element binding protein (CREB). These results suggest that (−)-sesamin has protective effects on both habit learning memory and spatial memory deficits via the dopaminergic neurons and NMDA receptor-ERK1/2-CREB system in MPTP-lesioned mouse model of PD, respectively. Therefore, (−)-sesamin may serve as an adjuvant phytonutrient for memory deficits in PD patients.
Go to : 
References
(1). Mayeux R.Annu. Rev. Neurosci. 2003. 26:81–104.

(2). Buter T. C.., van den Hout A.., Matthews F. E.., Larsen J. P.., Brayne C.., Aarsland D.Neurology. 2008. 70:1017–1022.
(3). Williams-Gray C. H.., Foltynie T.., Brayne C. E.., Robbins T. W.., Barker R. A.Brain. 2007. 130:1787–1798.
(4). Da Cunha C.., Angelucci M. E. M.., Canteras N. S.., Wonnacott S.., Takahashi R. N.Cell. Mol. Neurobiol. 2002. 22:227–237.
(5). Shi L.., Adams M. M.., Long A.., Carter C. C.., Bennett C.., Sonntag W. E.., Nicolle M. M.., Robbins M.., D'Agostino R.., Brunso-Bechtolda J. K.Radiat. Res. 2006. 166:892–899.
(6). Wang H. M.., Cheng K. C.., Lin C. J.., Hsu S. W.., Fang W. C.., Hsu T. F.., Chiu C. C.., Chang H. W.., Hsu C. H.., Lee A. Y.Cancer Sci. 2010. 101:2612–2620.
(7). Han A. R.., Kim H. J.., Shin M.., Hong M.., Kim Y. S.., Bae H.Chem. Biodivers. 2008. 5:346–351.
(8). Peñalvo J. L.., Hopia A.., Adlercreutz H.Eur. J. Nutr. 2006. 45:439–444.
(9). Park H. J.., Zhao T. T.., Lee K. S.., Lee S. H.., Shin K. S.., Park K. H.., Choi H. S.., Lee M. K.Neurochem. Int. 2015. 83–84:19–27.
(10). Fujikawa T.., Kanada N.., Shimada A.., Ogata M.., Suzuki I.., Hayashi I.., Nakashima K.Biol. Pharm. Bull. 2005. 28:169–172.
(11). Zhang M.., Lee H. J.., Park K. H.., Park H. J.., Choi H. S.., Lim S. C.., Lee M. K.Neuropharmacology. 2012. 62:2219–2226.
(12). Li C. Y.., Chow T. J.., Wu T. S. J.Nat. Prod. 2005. 68:1622–1624.
(13). Schober A.Cell Tissue Res. 2004. 318:215–224.
(14). Roghani M.., Baluchnejadnojarad T.Basic Clin. Neurosci. 2010. 1:52–55.
(15). Izurieta-Sánchez P.., Sarre S.., Ebinger G.., Michotte Y.Eur. J. Pharmacol. 1998. 353:33–42.
(16). Shin K. S.., Choi H. S.., Zhao T. T.., Suh K. H.., Kwon I. H.., Choi S. O.., Lee M. K.Arch. Pharm. Res. 2013. 36:759–767.
(17). Kumar B.., Kuhad A.., Chopra K.Psychopharmacology. 2011. 214:819–828.
(18). El Massioui N.., Delatour B. V.Neurosci. Res. Comm. 1997. 21:103–111.
(19). Matsumoto N.., Hanakawa T.., Maki S.., Graybiel A. M.., Kimura M. J.Neurophysiol. 1999. 82:978–98.
(20). Bannerman D. M.., Good M. A.., Butcher S. P.., Ramsay M.., Morris R. G.Nature. 1995. 378:182–186.
(21). Krapivinsky G.., Krapivinsky L.., Manasian L.., Ivanov A.., Tyzio R.., Pellegrino C.., Ben-Ari Y.., Clapham D. E.., Medina I.Neuron. 2003. 40:775–784.
(22). Carlezon Jr W. A.., Duman R. S.., Nestler E. J.Trends Neurosci. 2005. 28:436–445.
(23). Heikkila R. E.., Hess A.., Duvoisin R. C.Science. 1984. 224:1451–1453.
Go to :
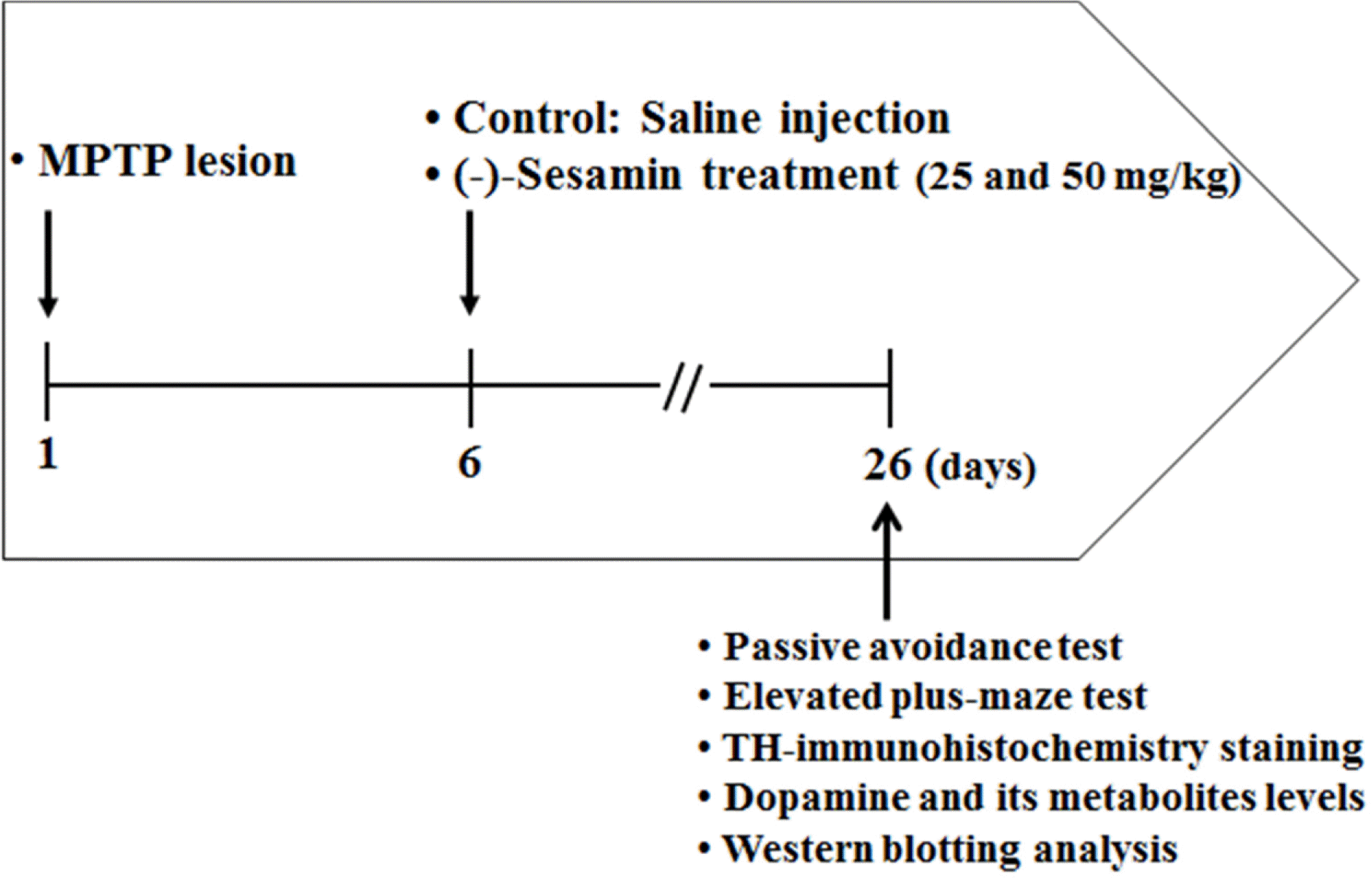 | Fig. 1.Experimental design. Mice (C57BL/6, male, 20 − 25 g) were divided into 4 groups (8 – 10 animals and lesions) and PD models were established with by MPTP injection (30 mg/kg, daily for 5 days, i.p.). Five days after lesion formation, MPTP-lesioned groups were treated with (−)-sesamin (25 and 50 mg/kg) orally for 21 days. After the final treatments, all mice were subjected to behavioral tests. The mice were then sacrificed to obtain brain tissues for biochemical, western blotting and immune histochemical analyses. |
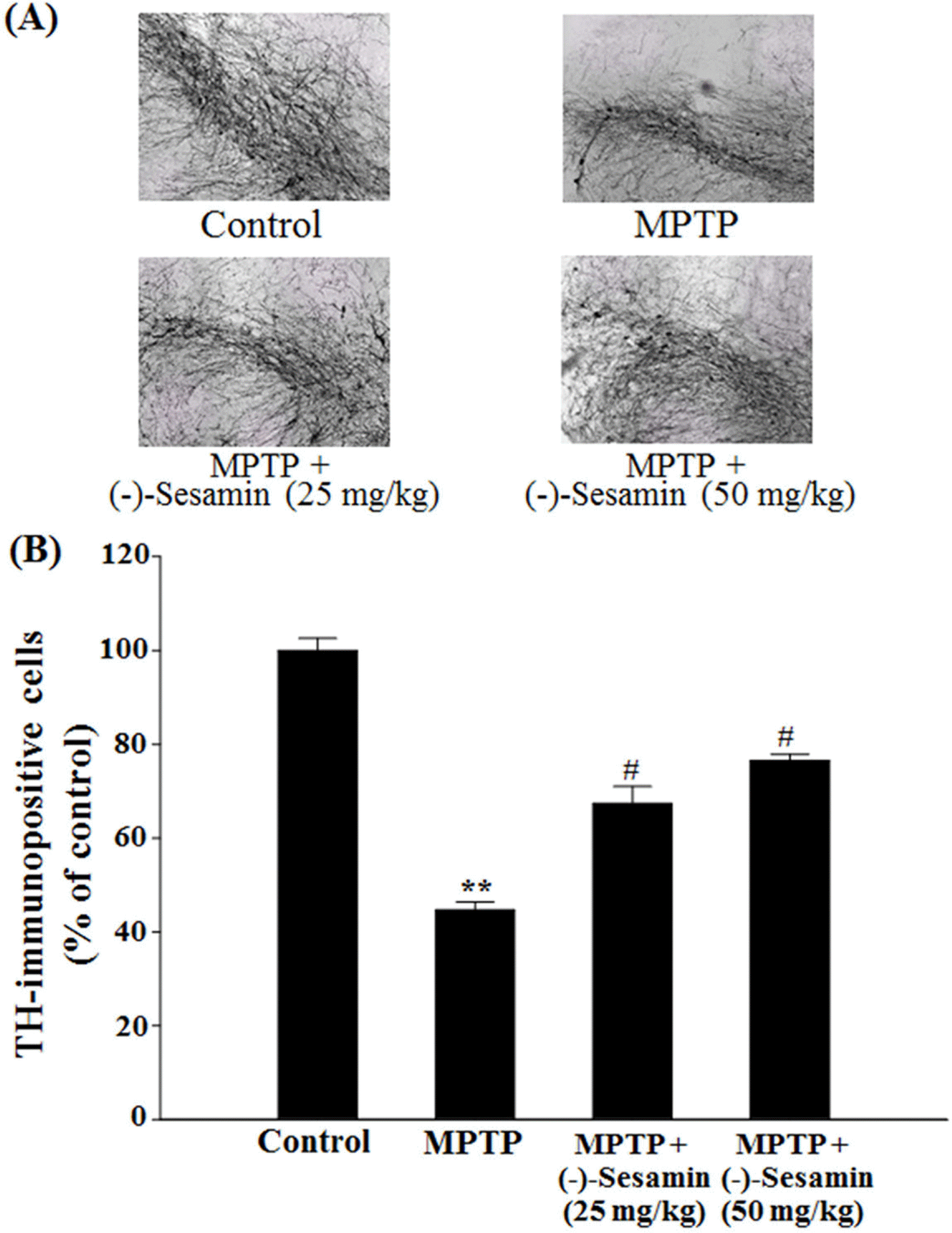 | Fig. 2.Representative photographs illustrating the effects of (−)-sesamin on TH-immunohistochemistry (A) and the number of TH-immunopositive cells (B) in the substantia nigra. Mice (C57BL/ 6, male, 20 – 25 g) were orally treated with (−)-sesamin (25 or 50 mg/kg) for 21 days after the 5 days of MPTP injections (30 mg/ kg, i.p.). The control group was treated with 0.9% saline. (A) Immunoblots of lysates from the brain were probed with TH antibodies, and the TH-immunopositive neuronal cells were measured as described under the Experimental section. (B) The number of TH-immunopositive cells was counted in the substantia nigra and was expressed as a percentage of the control groups. The results are expressed as the means ± S.E.M. (8 − 10 animals per group). ∗∗P < 0.01 compared with the control group; #P < 0.05 compared with the MPTP-lesioned group (one-way ANOVA followed by Tukey's test). |
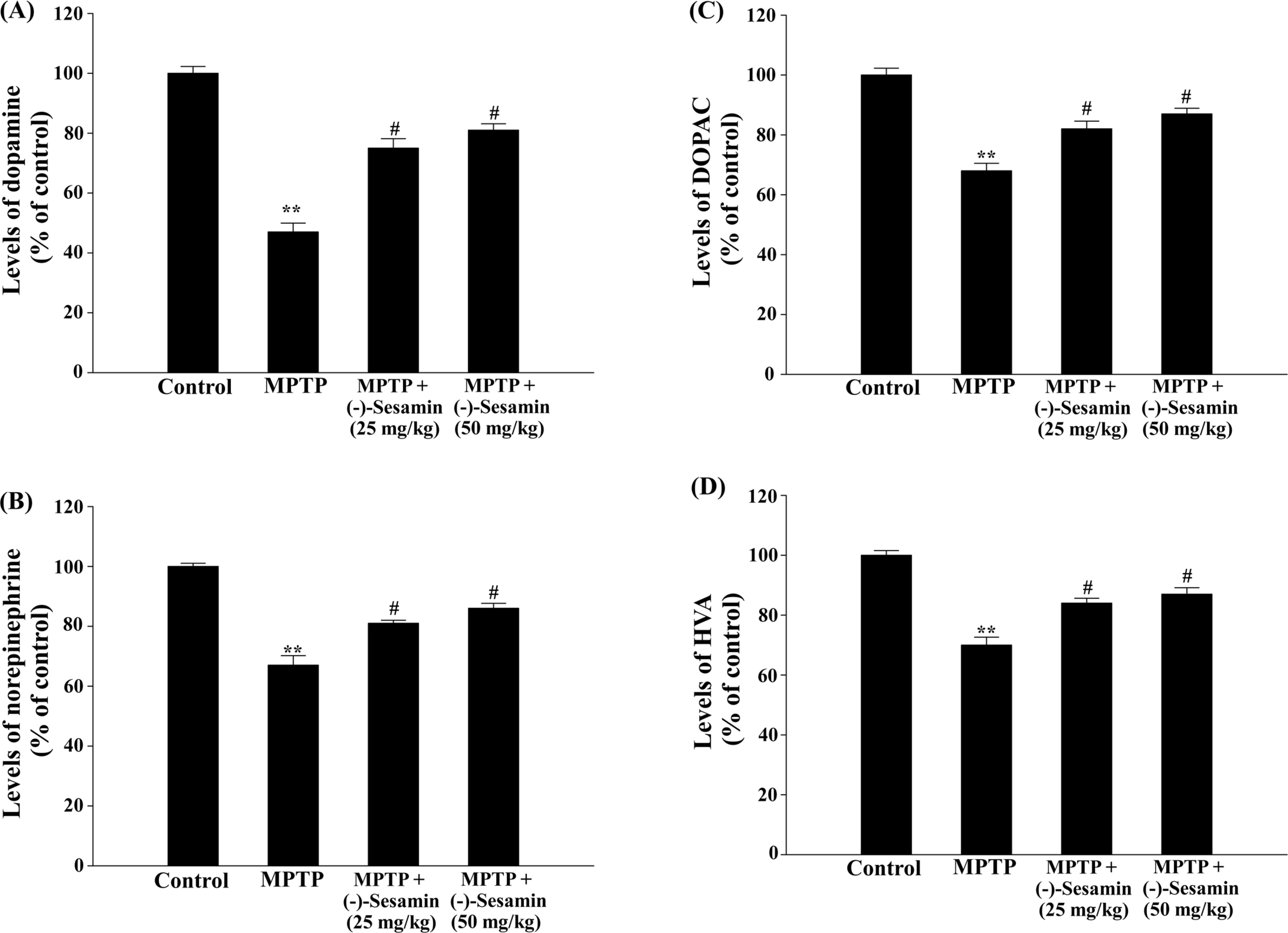 | Fig. 3.Effects of (−)-sesamin on dopamine (A), norepinephrine (B), DOPAC (C) and HVA (D) levels in the striatum. Mice (C57BL/6, male, 20 – 25 g) were orally treated with (25 or 50 mg/kg) for 21 days after the 5 days of MPTP injections (30 mg/kg, i.p.). The control group was treated with 0.9% saline. The control levels (ng/mg tissue) of dopamine (A), norepinephrine (B), DOPAC (C) and HVA (D) were 8.01 ± 0.71, 5.34 ± 0.61, 3.15 ± 0.49, and 1.96 ± 0.52, which were determined by HPLC analysis. The results are expressed as the means ± S.E.M. (8 − 10 animals per group). ∗∗P < 0.01compared with the control group; #P < 0.05 compared with the MPTP-lesioned group (one-way ANOVA followed by Tukey's test). |
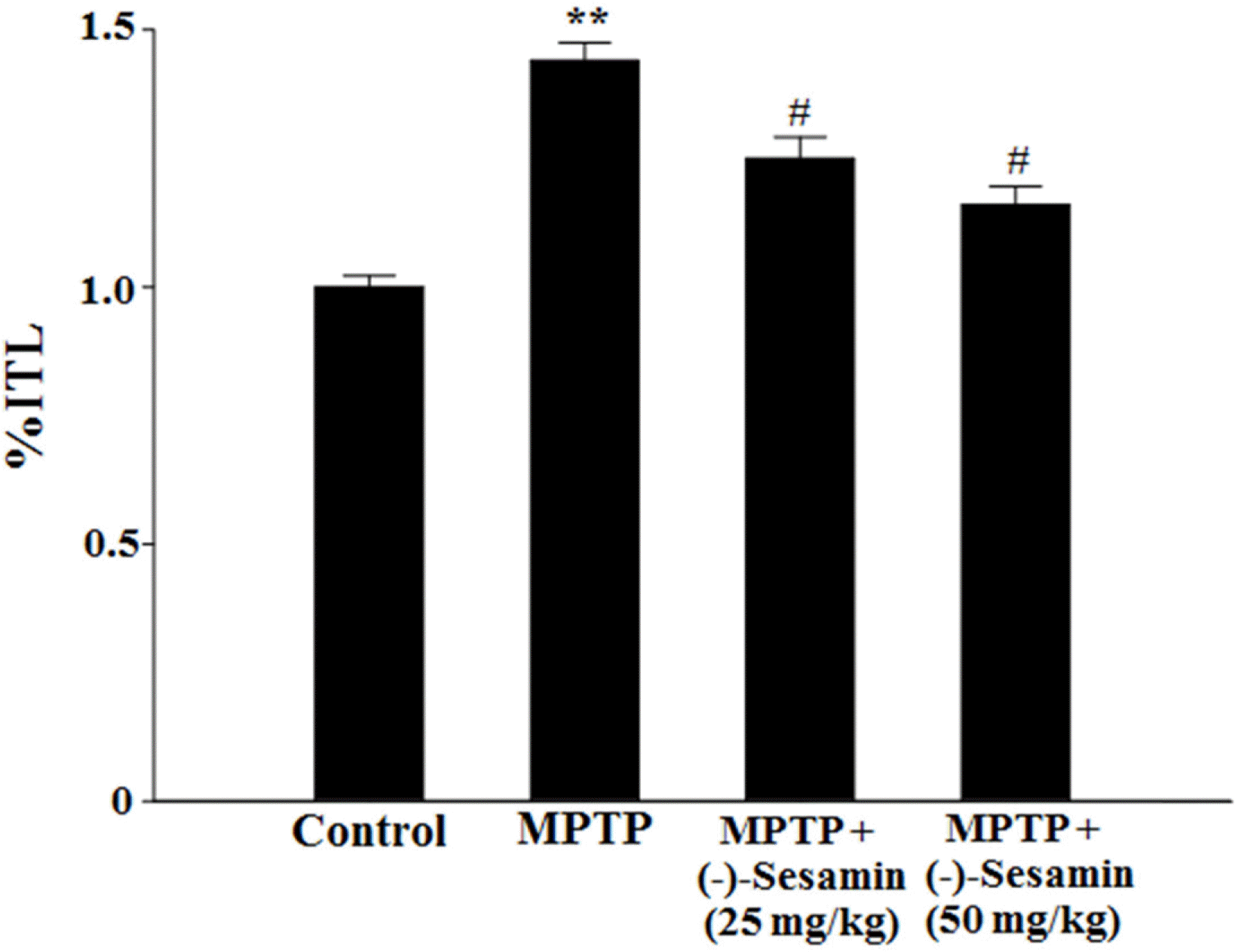 | Fig. 4.Effects of (−)-sesamin on transfer latency time (%ITL) in the elevate plus-maze test. Mice (C57BL/6, male, 20 – 25 g) were orally treated with (−)-sesamin (25 or 50 mg/kg) for 21 days after the 5 days of MPTP injections (30 mg/kg, i.p.). The control group was treated with 0.9% saline. After the 24-h of initial transfer latency test, the retention transfer latency time was noted and expressed as percentage of the initial transfer latency time (%ITL). The results are expressed as the means ± S.E.M. (8 − 10 animals per group). ∗∗P < 0.01 compared with the control group; #P < 0.05 compared with the MPTP-lesioned group (one-way ANOVA followed by Tukey's test). |
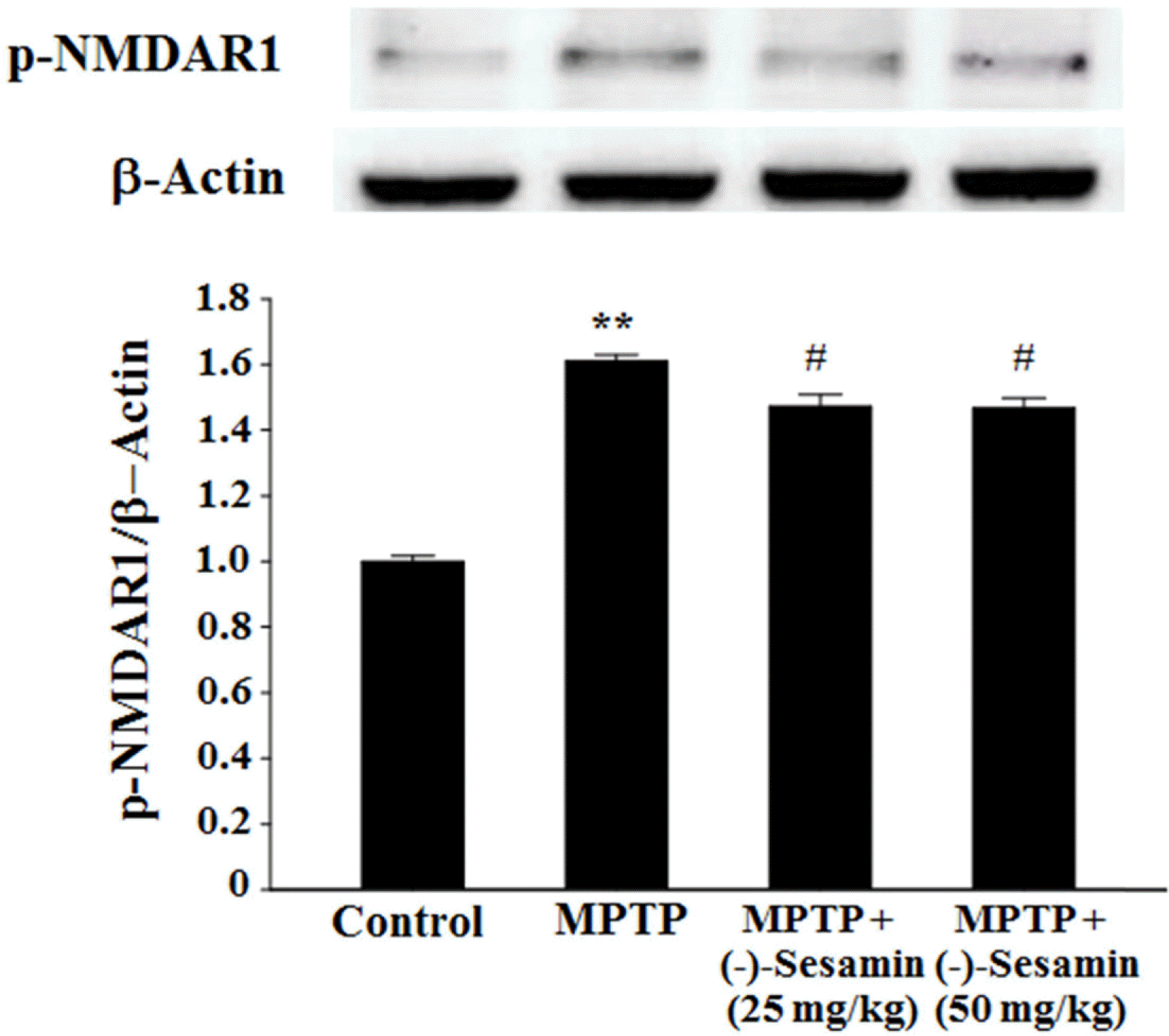 | Fig. 5.Effects of (−)-sesamin on phosphorylation of NMDAR1 in the hippocampus. Mice (C57BL/6, male, 20 – 25 g) were orally treated with (−)-sesamin (25 or 50 mg/kg) for 21 days after the 5 days of MPTP injections (30 mg/kg, i.p.). The control group was treated with 0.9% saline. Immunoblots of lysates from the mice were probed with phospho-NMDAR1 antibody, and the phosphorylation of NMDAR1 (Ser 897) (p-NMDAR1) and β-actin were analyzed by western blot analysis. The value of the relative density ratio of p-NMDAR1/β-actin is expressed in arbitrary units. The results are expressed as the means ± S.E.M. (8 − 10 animals per group). ∗∗P < 0.01 compared with the control group; #P < 0.05 compared with the MPTP group (one-way ANOVA followed by Tukey's test). |
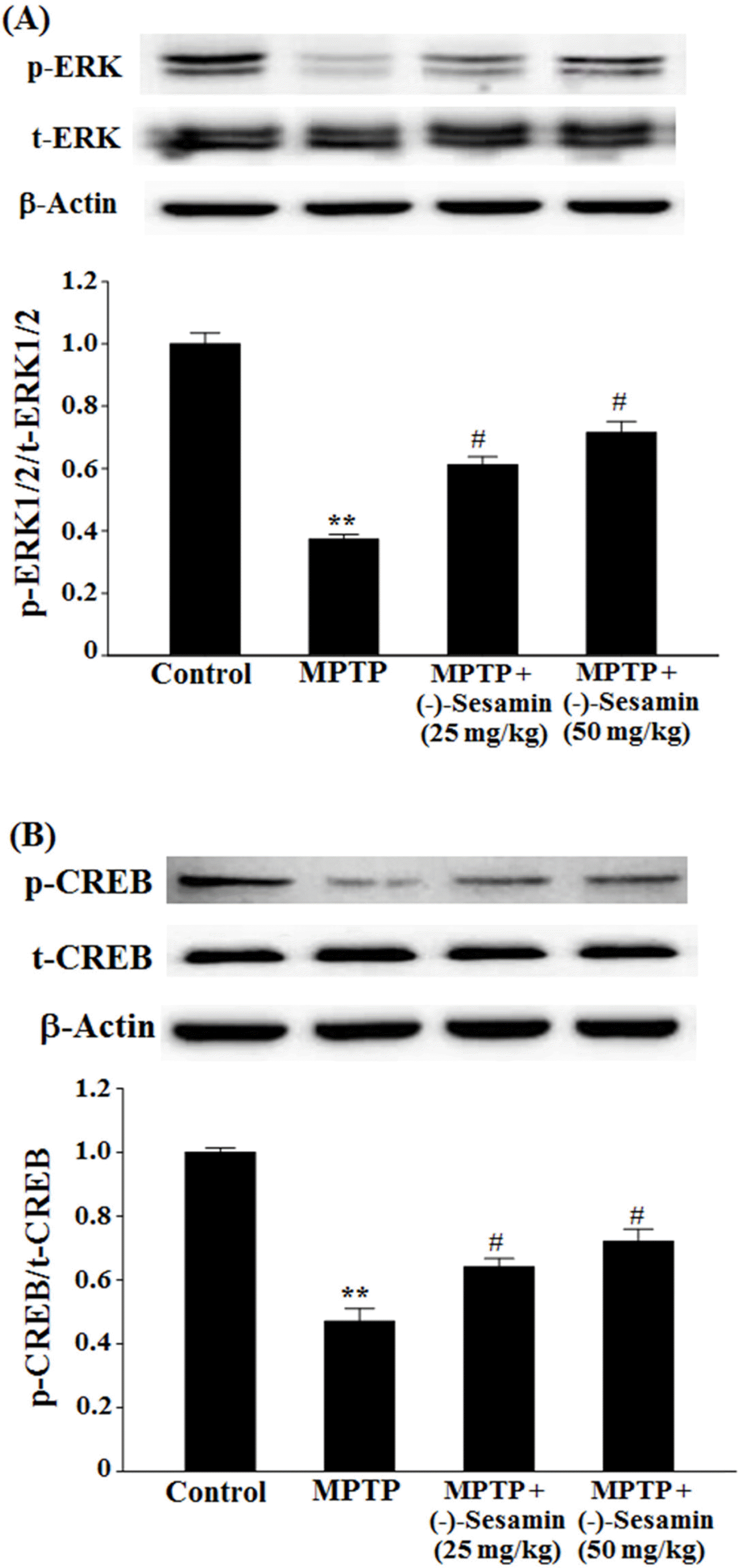 | Fig. 6.Effects of (−)-sesamin on phosphorylation of ERK1/2 (A) and CREB (B) in the hippocampus. Mice (C57BL/6, male, 20 – 25 g) were orally treated with (−)-sesamin (25 or 50 mg/kg) for 21 days after the 5 days of MPTP injections (30 mg/kg, i.p.). The control group was treated with 0.9% saline. Immunoblots of lysates from the mice were probed with phospho-ERK1/2 antibody, and the phosphorylation of ERK1/2 (Thr 202/Tyr 204) (p-ERK1/2), total ERK1/2 (t-ERK1/2) and β-actin were analyzed by western blot analysis. The value of the relative density ratio of p-ERK1/2/ t-ERK1/2 is expressed in arbitrary units. The results are expressed as the means ± S.E.M. (8 − 10 animals per group). ∗∗P < 0.01 compared with control group; #P < 0.05 compared with MPTP group (one-way ANOVA followed by Tukey's test). |
Table 1.
Effects of (−)-sesamin on latency time in the passive avoidance test
| Latency time (s) | ||
|---|---|---|
| Initial trial | Retention trial | |
| Control | 21.6 ± 1.68 | 130.4 ± 19.3 |
| MPTP | 17.9 ± 1.97 | 578.3 ± 16.4∗∗ |
| MPTP + (−)-Sesamin (25 mg/kg) | 18.5 ± 2.76 | 109.8 ± 18.5 |
| MPTP + (−)-Sesamin (50 mg/kg) | 21.7 ± 3.01 | 117.6 ± 16.3# |
Mice (C57BL/6, male, 20 – 25 g) were orally treated with (−)-sesamin (25 or 50 mg/kg) for 21 days after the 5 days of MPTP injections (30 mg/kg, i.p.). The control group was treated with 0.9% saline. After the final treatments, all mice were subjected to the passive avoidance test. The results are expressed as the means ± S.E.M. (8 – 10 animals per group).
 XML Download
XML Download