

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Alcohol-induced liver damage is a major health problem worldwide. Liver injury secondary to alcohol consumption is the result of complex processes involving multiple organ systems including intestinal barrier function, the innate immune system, and adipose tissue. Initial exposure to alcohol causes imbalances in the hepatic redox status and reactions of alcohol metabolites that are toxic to hepatocyte proteins and lipid membranes. Prolonged alcohol intake may impair liver function due to the accumulation of fatty acids and the generation of reactive oxygen species (ROS) by the cytochrome P450 system, which induce liver inflammation and fibrogenesis.12 Intracellular ROS production plays a crucial role in the development of alcohol-induced liver damage by causing lipid peroxidation of cellular membranes and protein and DNA oxidation. Ethanol metabolism is not only directly involved in the generation of ROS, but is also linked to depletion of cellular anti-oxidants. Glutathione (GSH) is a crucial cellular anti-oxidant responsible for limiting the toxicity of ethanol as well as many other toxic chemicals. Chronic ethanol exposure diminishes GSH levels and down-regulates anti-oxidative enzymes such as superoxide dismutase (SOD) and glutathione peroxidase (GPx).34 The activation of the ethanol-inducible cytochrome P450 2E1 (CYP2E1) system causes increased generation of oxygen- and ethanol-derived free radicals. CYP2E1 is capable of producing the superoxide anion, hydrogen peroxide, and ethanol-derived hydroxyethyl free radicals, as well as highly toxic metabolites derived from CYP2E1 substrate xenobiotics.5 In addition, ROS may be derived from activated Kupffer cells in the liver along with tumor necrosis factor alpha (TNF-α), which is one of the principal mediators of the inflammatory response in mammals. Accumulated evidence supports the hypothesis that chronic alcohol exposure disturbs intestinal barrier function and that gut-derived endotoxins play a critical role in the production of ROS and TNF-α by Kupffer cells.6 Down-regulation of TNF-α production by anti-oxidants is paralleled by attenuated hepatic lipid peroxidation, steatosis, and liver injury.7 Fatty liver is another phenomenon that occurs in ethanol-induced liver damage. Hepatosteatosis caused by ethanol consumption can be associated with many factors, including the redox status of liver cells, impaired transportation of synthesized lipids, inhibition of fatty acid oxidation, and abnormal enhancement of lipogenesis.8
Therapeutic strategies for alcohol-induced liver damage target ROS generation, cytokine overproduction, and the translocation of endotoxins from the gut.2 Diets or food supplements rich in natural anti-oxidants could be promising candidates for the prevention of alcohol-induced liver damage. For example, silymarin, a mixture of flavonolignans extracted from milk thistle (Silibum marianum), protects against ethanol-induced liver injury by promoting anti-oxidation.9 One model system for alcohol-induced liver damage uses rats fed the Lieber-DeCarli liquid diet, which is nutritionally balanced between the control and ethanol groups. Rats fed the Lieber-DeCarli ethanol diet for 4 weeks developed significant liver lesions, steatosis, apoptosis, CYP2E1 induction, generation of free radicals, and alterations of defense mechanisms against oxidative stress.10
The root of the Paeonia species is widely used in Asian countries as a traditional medicine. The root of Paeonia anomala L. (Paeoniaceae) is used in Mongolian traditional medicine to treat lower abdominal pain, kidney diseases, and abnormal blood clotting.11 The fruit component (seedcases without seeds) of P. anomala (Sogoon sav in Mongolian) is a famous traditional medicine in Mongolia and is used to treat gynecological diseases, kidney disorders, and bladder inflammation12; however, the usage methods are poorly reported in the literature.
In the present study, the hepatoprotective effect of a fruit extract of P. anomala (FEPA) against chronic alcohol-induced liver damage was evaluated in rats fed the Lieber-DeCarli liquid diet for 5 weeks. FEPA attenuated ethanol-induced liver injury by inhibiting the development of fatty liver, decreasing the production of pro-inflammatory cytokines, and increasing the anti-oxidative capacity.
Experimental
Plant extract
Fruits (seedcases without seeds) of P. anomala were collected in Bayanchandmani sum, Tov province, which is 70 km northwest of Ulaanbaatar, Mongolia, in October 2011 and identified by Dr. C. Sanchir at the Institute of Botany of the Mongolian Academy of Sciences. A voucher specimen (2011/60) was deposited in the Flora and Plant Systematic Laboratory, Institute of Botany, Mongolian Academy of Sciences. FEPA was prepared as mentioned previously and its chemical constituents have been reported.13
Animals and experimental design
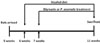
Five-week-old male Sprague-Dawley male rats with a body weight (bw) of 140 – 160 g were obtained from Orient Bio, Daegu, Republic of Korea. Animal care and handling were performed following the guidelines of the Institutional Animal Care and Use Committee of the Korea Institute of Science and Technology. The rats were kept under the standard conditions of the animal house with a 12:12 light:dark cycle at a temperature of 22 ± 2 ℃. They had free access to food and water for 1 week before the experiment. The rats were divided into six groups, each containing seven rats. They were housed individually and fed 250 mL/kg bw/day Lieber-DeCarli control or ethanol diets (Dyets Inc., Bethlehem, PA, USA) for 1 week. The ethanol dose in the alcohol-treated groups was 11 g/kg bw/day. One week later, FEPA (50, 25, and 10 mg/kg bw/day) or the reference control silymarin (25 mg/kg bw/day) was dissolved in ethanol, mixed with the Lieber-DeCarli ethanol diet, and fed to the experimental animals for 4 weeks (Fig. 1). At the end of the experiment, the animals were fasted overnight and sacrificed using diethyl ether. Blood was collected in EDTA-containing tubes (BD Science, Franklin Lakes, NJ, USA) and liver tissue was collected after perfusion with 0.15% KCl solution.
Serum enzyme assays
The sera from blood samples were collected by centrifugation at 2,580 × g for 10 min and stored at −80 ℃ until the assay. Serum activity levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using NADH enzyme colorimetric kits (Young-Dong Pharm. Co., Yongin, Korea).
Measurement of hepatic TNF-α, triglycerides (TGs), and the anti-oxidative capacity
Liver tissue was homogenized in ice-cold liver homogenization buffer (0.25M K2HPO4/KH2PO4 and 0.15 M KCl, pH 7.25) and centrifuged at 2,580 × g for 20 min. The supernatant was transferred into a new tube and centrifuged at 100,900 × g for 30 min. The resulting supernatant was used to measure TNF-α, TGs, and the anti-oxidative capacity. TNF-α levels were detected using the commercial TNF-α rat enzyme-linked immunosorbent assay kit (Abcam Inc., Cambridge, MA, USA) according to the manufacturer's instructions. The hepatic TG content was measured using the TG colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA). The oxygen radical anti-oxidant capacity (ORAC) assay kit (Cell Biolabs Inc., San Diego, CA, USA) was used to determine the total hepatic anti-oxidant capacity.
Lipid peroxidation and CYP2E1 enzyme assays
Pellets obtained by the centrifugation of liver tissue at 100,900 × g for 30 min were resuspended in ice-cold liver homogenization buffer and sonicated for 4 sec at an amplitude of 10%. After centrifugation at 14,980 × g, the supernatant was used for lipid peroxidation and CYP2E1 enzyme assays. Lipid peroxidation was assessed by measuring the formation of thiobarbituric acid-reactive substances (TBARS) according to previously described methods.14 CYP2E1 enzyme activity was quantified calorimetrically using previously reported methods15 with minor modifications. Briefly, 405 µL of liver homogenization buffer, 10 µL of 5 mM p-nitrophenol, 25 µL of 66 mM MgCl2, and 20 µL of 5 mM NADPH were mixed in eppendorf tubes and pre-incubated for 30 min. Then, 40 µL of the experimental sample or the p-nitrocatechol standard was added to the tubes and incubated for 30 min at 37 ℃, after which 100 µL of 20% trichloroacetic acid was added and the tubes were incubated on ice for 30 min. The tubes were centrifuged at 10,000 × g for 5 min, and 100 µL of the supernatant was transferred to a 96-well plate containing 50 µL of 2M NaOH. CYP2E1-catalyzed p-nitrophenol hydroxylation was determined by measuring absorbance at 535 nm. All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).
Determination of hepatic total glutathione (t-GSH) and anti-oxidative enzyme activity
SOD and GPx activity levels were measured in supernatants obtained after the centrifugation of liver tissue homogenates in icecold buffer containing 0.25M K2HPO4/KH2PO4 and 0.15 M KCl (pH 7.25) at 100,900 × g for 30 min. SOD activity was measured by the reduction of nitrobluetetrazolium by the xanthine-xantine oxidase system, and GPx activity was determined by NADPH oxidation. Hepatic t-GSH (GSH and glutathione disulfide) activity was assessed in liver extracts incubated in 5% metaphosphoric acid using enzymatic recycling methods as previously described.14 All chemicals were purchased from Sigma-Aldrich. Protein concentrations in the homogenates were quantified using a Bradford assay with the Bio-Rad protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Result and Discussion
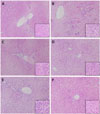
Liver sections of rats fed the alcohol diet showed that the radial arrangements of the hepatic plates were often disrupted, and hepatocytes became foamy and were filled with vacuole-like spaces due to retained fatty acids (Fig. 2). However, FEPA treatment preserved the radial arrangement and inhibited the formation of fatty acidfilled vacuoles in the liver, demonstrating the hepatoprotective effect of FEPA against alcohol-induced liver damage.
Ethanol administration for 5 weeks decreased the bws of rats and increased the liver-to-bw ratio. Only the group treated with ethanol alone showed a statistically significant increase in the liver-to-bw ratio compared with the vehicle group (Table 1). This may be a consequence of fatty acid deposition and/or impaired proteasome activity in the liver.16 FEPA treatment at doses of 25 and 10 mg/kg bw/day decreased the liver-to-bw ratio compared with the ethanol-only group; however, statistical significance was not observed.
Liver damage was biochemically evaluated by assessing the activity levels of serum enzymes (ALT and AST), which were significantly increased in rats fed the ethanol diet (Table 2). Ethanol treatment elevated the AST level by 2.5-fold and the ALT level by 4-fold compared with the vehicle group. This result showed that alcohol consumption for 5 weeks caused liver damage, as expected. However, treatment with FEPA and silymarin attenuated the increases in the serum levels of AST and ALT. Even the lowest doses of FEPA decreased the levels of serum enzymes more potently than the reference control silymarin. One possible explanation for this observation is that the activity of FEPA is a cumulative effect of all the components of the extract because FEPA also contains the toxic compound gnetin-H. In our laboratory, IC50 values (the concentration that inhibits 50% of cell proliferation) for gnetin-H after 24 hours of treatment were 27 µM for HepG2 cells and 5 µM for Hepa1c1c7 cells. Thus, at the higher dose of FEPA, the adverse effect of gnetin-H affected the hepatoprotective effect of this extract.
We further assessed the hepatoprotective effects of FEPA against TG accumulation, TNF-α production, and oxidative stress. Increased fatty acid deposition, abnormal cytokine production, and oxidative stress play a pivotal role in the initiation of alcohol-induced liver damage. TNF-α is believed to be a crucial cytokine in alcohol-induced liver damage because it mediates inflammatory responses, steatosis, and cell death.17 Alcohol intake promotes hepatic inflammation by increasing the translocation of gut-derived endotoxins to the portal circulation and activating Kupffer cells, which then release TNF-α as well as ROS. Released TNF-α can induce cell death by apoptosis, and apoptotic hepatocytes can stimulate Kupffer cells to produce more TNF-α.18 Thus, inhibition of TNF-α production by Kupffer cells correlates with the amelioration of alcohol-induced liver damage.19 In our experiment, TNF-α production in the liver was significantly increased by alcohol intake; levels were 59.2 pg/mg protein in the alcohol-treated group vs. 35.1 pg/mg protein in the vehicle group (Table 3). This increased TNF-α production was significantly reduced to 39.1 and 45.6 pg/mg protein by treatment with 10 and 25 mg/kg bw/day FEPA, respectively. Silymarin had a weak inhibitory effect on alcohol-induced production of TNF-α.
One of the early processes in the development of alcohol-induced liver damage is fatty acid accumulation in the liver. The mechanism underlying fatty liver formation by ethanol is quite complex and involves impaired transportation of lipids, decreased fatty acid oxidation, and abnormally increased lipid synthesis. In the initial stages of alcohol-induced liver damage, ethanol metabolism by alcohol dehydrogenase results in enhanced levels of NADH, which can inhibit the NAD+-requiring tricarboxylic acid cycle and β-oxidation of fatty acids.6 However, fatty liver formation was not attenuated by changes in the redox status in chronically ethanol-treated animals,20 and anti-oxidant treatment was not found to be an effective treatment for alcohol-induced steatosis.21 In this study, ethanol administration induced significant accumulation of TGs in the liver; TG levels were 3-fold higher in the ethanol-only group than in the vehicle group; however, this accumulation was attenuated by all doses of FEPA (Table 3).
Alcohol exposure induces oxidative stress via many pathways and results in the peroxidation of lipids, proteins, and DNA. Many pathways for this phenomenon have been reported, including the production of reactive products from acetaldehyde produced from ethanol oxidation, damage to mitochondria resulting in decreased ATP production, toxic products generated by ethanolinduced CYP2E1 activity, the effects of alcohol on antioxidative enzymes and small anti-oxidants, particularly mitochondrial and cytosolic GSH, ethanol-induced hypoxia, and 1-hydroxyethyl radical formation.3
The microsomal ethanol-oxidizing system is a minor pathway for ethanol metabolism and its induction has been observed in alcoholics. CYP2E1 is a major component of cytochrome P450, which is inducible by chronic ethanol intake and catalyzes the oxidation of ethanol to acetaldehyde. CYP2E1 also promotes ethanol oxidation to convert ethanol into the 1-hydroxyethyl radical, and this effect is inhibited by SOD.24 A decrease in CYP2E1 induction is associated with amelioration of alcohol-induced liver injury.3 In our experiment, liver CYP2E1 enzyme activity was 2.5-fold higher in the alcohol-treated group than in the vehicle group; however, co-treatment with FEPA attenuated this increase. Upon administration of the lowest dose of FEPA (10 mg/kg bw/day), CYP2E1 activity was similar to that in the vehicle group (Table 4).
Moreover, hepatic lipid peroxidation is often assessed in animal models as a biomarker of hepatic oxidative stress.22 In our experiment, lipid peroxidation was evaluated in liver homogenates by assessing the TBARS content. Alcohol administration significantly increased the hepatic TBARS content, but both FEPA and silymarin attenuated hepatic lipid peroxidation. FEPA at a dose of 10 mg/kg bw/day showed the most potent protective effect against alcohol-induced hepatic lipid peroxidation. An ORAC assay was performed to evaluate the hepatic anti-oxidative capacity. Alcohol consumption significantly decreased the hepatic anti-oxidative capacity; however, administration of low doses of FEPA (25 and 10 mg/kg bw/day) led to recovery of the hepatic anti-oxidative capacity.
Furthermore, chronic alcohol administration is a cause of the decreased anti-oxidative defense capacity, as reflected by levels of GSH and anti-oxidative enzymes such as SOD and GPx.3 Studies employing a 20% alcohol water model found decreases in SOD activity in the liver.23 Based on our results, alcohol administration reduced SOD activity in the liver, although the difference between the vehicle and ethanol-only groups was not statistically significant. GPx is an essential anti-oxidative enzyme that removes hydrogen peroxide by reacting with GSH, glutathione reductase, and NADPH.24 In this experiment, the level of the redox status biomarker t-GSH was reduced to 13.65 nM/mg protein in the livers of rats fed the ethanol diet, but this was recovered to 16.23 and 15.41 nM/mg protein in rats treated with 10 mg/kg bw/day FEPA and silymarin, respectively. From these results, we conclude that FEPA contributes to the recovery of alcohol-induced depletion of anti-oxidative enzyme activities and GSH levels. The activity levels of SOD and GPx, which are crucial anti-oxidative enzymes in the liver, were determined to assess the hepatic anti-oxidative defense system. GPx activity was reduced in the liver homogenates of rats fed ethanol. All doses of FEPA and silymarin slightly increased GPx activity compared with the alcohol-only group, but the effect of FEPA was not dose-dependent. Ethanol administration slightly decreased hepatic SOD activity, but silymarin and all doses of FEPA attenuated the decrease in SOD activity, although statistical significance was not observed. Our hypothesis posits that the anti-oxidative components of FEPA contribute to its hepatoprotective effects because anti-oxidants can scavenge ROS and decrease oxidative stress in the liver. However, FEPA may have other components that ameliorate hepatosteatosis because anti-oxidants alone cannot protect the liver from steatosis.21 The protective effect of FEPA against alcohol-induced liver damage was reversely correlated with the dosage of FEPA. The toxic component of FEPA, gnetin-H, may have an adverse effect on hepatoprotection by FEPA; thus eliminating gnetin-H from FEPA is essential for the development of further FEPA-based technologies and products.
In conclusion, FEPA exerts a hepatoprotective effect against alcohol-induced liver damage by inhibiting abnormal TNF-α production, diminishing hepatic TG levels, decreasing lipid peroxidation, and enhancing the activities of anti-oxidative enzymes and non-enzymatic radical scavengers. Considering these results, FEPA could be a promising candidate for the development of a functional food with a hepatoprotective effect against chronic alcohol consumption. Further studies on extract standardization and clinical trials are needed to develop a dietary supplement to enhance liver function.




 XML Download
XML Download