

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Oxidative stress occurs because of an imbalance between reactive oxygen species (ROS) production and the antioxidant defense system. ROS play a key role in oxidative stress and cause oxidative damage to lipids, proteins, and DNA either directly or by altering signaling pathways [1]. ROS, including hydrogen peroxide (H2O2), superoxide radical, hydroxyl radical, and peroxynitrite, can be generated either exogenously or endogenously from several factors such as the mitochondrial respiratory chain and cytosolic enzyme systems [23]. In particular, in many clinical situations, such as surgery, acute inflammatory processes, and anesthesia, ischemia/reperfusion (I/R) injury can occur and induce ROS production, which contributes to cellular dysfunction and the initiation of the apoptotic/necrotic cascade [34].
Autophagy, which is the mechanism that decreases ROS concentration and oxidative damage to biomolecules and organelles, is activated by ROS and reactive nitrogen species in response to oxidative stress [5]. Autophagy is an intracellular catabolic mechanism for degrading and recycling unnecessary or dysfunctional cellular components in response to various conditions [6]. Through autophagy, cells maintain their energy levels and synthesize structures required for survival [5]. ROS have been reported to be crucial inducers of autophagy, but the unexpected energetic requirement that develops during oxidative stress, in turn, overburdens the mitochondria and impairs its function. Moreover, ROS is generated at high concentrations in such situations, thereby suppressing the antioxidant role of autophagy and accelerating apoptosis [789].
Propofol (2,6-diisopropylphenol) is a widely used intravenous sedative-hypnotic agent for the induction and maintenance of general anesthesia, as well as for sedation in intensive care units. Some studies have demonstrated the antioxidant properties of propofol both in vitro [1011] and in vivo [12]. Propofol has a phenolic hydroxyl group, which is a structural characteristic similar to that of α-tocopherol (vitamin E), a type of endogenous antioxidant; accordingly, propofol presents antioxidant properties [13].
For protection against tissue damage from I/R injury, anesthetic preconditioning, which is defined as a brief exposure to anesthetic agents that protect the myocardium from the potentially fatal consequences of a subsequent prolonged period of myocardial I/R, has been increasingly used as a therapeutic strategy [14]. Propofol preconditioning (PPC) has been shown to protect the organs against I/R injury when propofol is administered before the ischemic period [1516]. The mechanisms underlying the protective effects of PPC have been shown to be the activation of adenosine triphosphate-sensitive potassium (KATP) channels, protein kinase C, protein tyrosine kinase, mitogen-activated protein kinase (MAPK), and protein kinase B (Akt) pathways [171819]. Such mechanisms may be related to the antioxidant effects of propofol because ROS are produced during I/R, and therefore, oxidative stress is closely related to I/R injury. However, the mechanisms underlying the effect of PPC on oxidative injury remain unclear. In this study, we used COS-7 cells (African green monkey kidney fibroblast-like cell line) to investigate whether PPC protects against cell damage caused by H2O2-induced oxidative stress and influences cellular autophagy.
MATERIALS AND METHODS
1. Chemicals and antibodies
Propofol, H2O2, and Hoechst 33342 were purchased from Sigma Chemicals (St Louis, MO, USA). The reagents 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide thiazolyl blue (MTT), acridine orange (AO), monodansylcadaverine (MDC), and 3-methyladenine (3-MA, class III phosphoinositide 3-kinase inhibitor) were obtained from Calbiochem (La Jolla, CA, USA). Antibodies used in the study were as follows: LC3-II (1:1,000) and Becline-1 (1:1,000) from Abcam, and p62 (1:1,000) and Atg5 (1:1,000) from Santa Cruz. Secondary antibodies against rabbit (1:5,000) and mouse (1:5,000) immunoglobulins were purchased from Bio-Rad (Richmond, CA, USA).
2. Cell cultures
The COS-7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The COS-7 cells were grown in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% inactivated fetal bovine serum (GIBCO) containing 500 µg/ml penicillin and 500 µg/ml streptomycin (GIBCO) and incubated at 37℃ in a humidified atmosphere of 95% air and 5% CO2. The medium was replaced every 3 days.
3. Treatment with propofol and H2O2
The propofol stock was prepared by dissolving it in dimethyl sulfoxide (DMSO) and was kept frozen at -4℃ until use. The stock was diluted to necessary concentrations by using DMEM when needed. The cells were grown to about 80% confluence and treated with H2O2 (400 µM) for 2 h with or without propofol. Prior to the addition of H2O2, the cells were exposed to propofol (100 µM) for 2 h. The cells grown in a medium containing an equivalent amount of DMSO without propofol and H2O2 served as the controls. The cells were divided into the following four groups: control, cells were incubated in normoxia (5% CO2, 21% O2, and 74% N2) for 24 h without propofol; H2O2, cells were exposed to H2O2 for 2 h; PPC + H2O2, cells pretreated with propofol for 2 h were exposed to H2O2; 3-MA + PPC + H2O2, cells pretreated with 3-MA (1 mM) for 1 h were exposed to propofol for 2 h and H2O2. 3-MA was used to identify whether the effect of PPC was mediated by autophagy, because 3-MA suppresses autophagy by blocking autophagosome formation.
4. Determination of cell viability by using MTT reduction
Cell viability was measured using the MTT assay, showing the mitochondrial activity of living cells. COS-7 cells (3 × 104) were seeded in 96-well plates. After drug treatment as indicated, the cells were incubated with 100 µl MTT (final concentration 0.5 mg/ml) for 4 h at 37℃. The reaction was terminated by adding 200 µl DMSO. The cells were exposed to propofol at various concentrations (0, 3, 30, 100, and 300 µM) for 2 h. To investigate the influence of H2O2 on COS-7 cell viability, the cells were treated with H2O2 at various concentrations (0, 50, 100, 200, and 400 µM) for 2 h. Cell viability was measured using an ELISA reader (Tecan, Männedorf, Switzerland) at 620 nm.
5. Fluorescence microscopy
After treatment with H2O2 (400 µM) for 2 h with or without propofol, the cells were stained with Hoechst 33342 (Molecular Probes, Eugene, OR, USA) and visualized under fluorescence microscopy to evaluate the morphological changes to the nuclei as a measure of apoptosis. To further evaluate the effect of propofol on the autophagy of COS-7 cells, we conducted autophagosome staining. Cells were grown on coverslips and treated with COS-7 cells. After 24 h, the cells were stained with 0.05 mM MDC, a selective fluorescent marker for autophagic vacuoles, at 37℃ for 1 h. The cellular fluorescence changes were observed under a fluorescence microscope (Axioskop, Carl Zeiss, Germany). For further detection of the acidic cellular compartment, we used AO, which emits bright red fluorescence in acidic vesicles but green fluorescence in the cytoplasm and nucleus. The cells were stained with 1 µg/ml AO for 15 min and washed with phosphate-buffered saline (PBS). Autophagic vesicle organelle (AVO) formation was observed under a confocal microscope (LSM 700, Carl Zeiss).
6. Flow cytometry analysis
The cells were seeded in 60-mm dishes at 70% confluence and incubated overnight. The propofol-pretreated cells were incubated for different periods according to the conditions. The harvested cells were washed with PBS containing 1% bovine serum albumin (BSA) and centrifuged at 2500 rpm for 10 min. The cells were then resuspended in ice-cold 95% ethanol with 0.5% Tween 20 to a final concentration of 75% ethanol. After 24 h, the fixed cells were washed in 1% BSA-PBS solution, resuspended in 1 ml PBS containing 40 µg/ml RNase A (Sigma), incubated at 4℃ for 30 min, and resuspended in 10 µg/ml propidium iodide solution (Sigma). The DNA contents were examined using a CYTOMICS FC500 flow cytometer (Beckman Coulter) and the data were analyzed using the MultiCycle software, which allowed simultaneous estimation of cell-cycle parameters and apoptosis.
7. Western blot analysis
The cells (2 × 106) were washed twice in ice-cold PBS, resuspended in 200 µl ice-cold solubilizing buffer (300 mM NaCl, 50 mM Tris-Cl [pH 7.6], 0.5% Triton X-100, 2 mM phenylmethylsulfonyl fluoride, 2 µl/ml aprotinin, and 2 µl/ml leupeptin) and incubated at 4℃ for 30 min. The lysates were centrifuged at 14,000 rpm for 15 min at 4℃. Protein concentrations of the cell lysates were determined using the Bradford protein assay (Bio-Rad), and 25 µg of proteins were resolved by using 10% SDS/PAGE. The gels were transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA) and reacted with appropriate primary antibodies. Immunostaining with secondary antibodies was detected using SuperSignal West Femto (Pierce, Rockford, IL, USA) enhanced chemiluminescence substrate and detected using AlphaImager HP (Alpha Innotech, Santa Clara, CA, USA).
8. Statistical analysis
The results were presented as the means ± standard deviation (SD). The study results were analyzed using analysis of variance followed by Tukey's post-hoc test. Simple pairwise comparisons were analyzed for significance by using Student's t-test. Statistical significance was defined as P < 0.05.
RESULTS
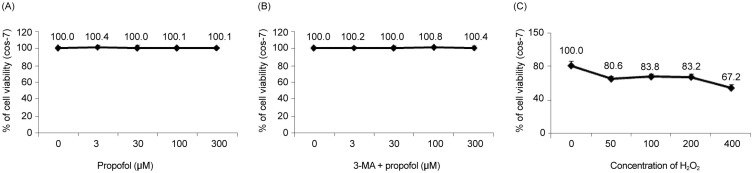
1. Propofol improved the decrease in H2O2-induced COS-7 cell viability
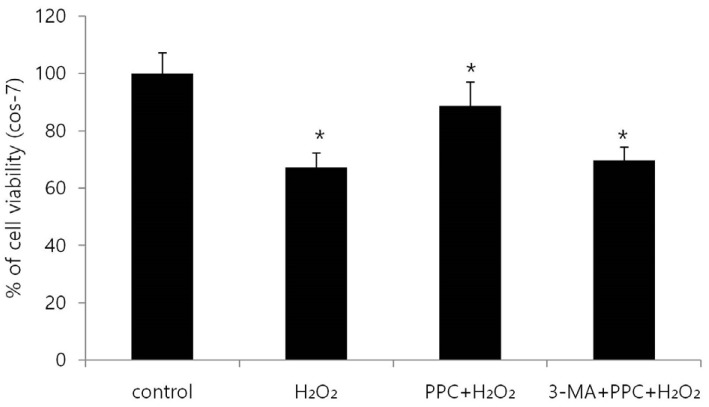
We examined the effects of propofol and H2O2 injury on COS-7 cell viability by measuring MTT reduction. Propofol treatment at various concentrations (0, 3, 30, 100, and 300 µM) produced no significant differences in COS-7 cell viability (Fig. 1A). No change in cell viability was observed when the cells were pretreated with 3-MA before propofol administration (Fig. 1B). However, the application of H2O2 to COS-7 cells at various concentrations (0, 50, 100, 200, and 400 µM) for 24 h resulted in a decrease in cell viability (Fig. 1C). As shown in Fig. 2, compared to the control group, the H2O2 group showed a significant decrease in cell viability (P < 0.05), which was improved by PPC (100 µM). The 3-MA (1 mM) treatment 1 h before H2O2 exposure inhibited the protective effect of PPC and increased H2O2-induced COS-7 cell apoptosis (Fig. 2).
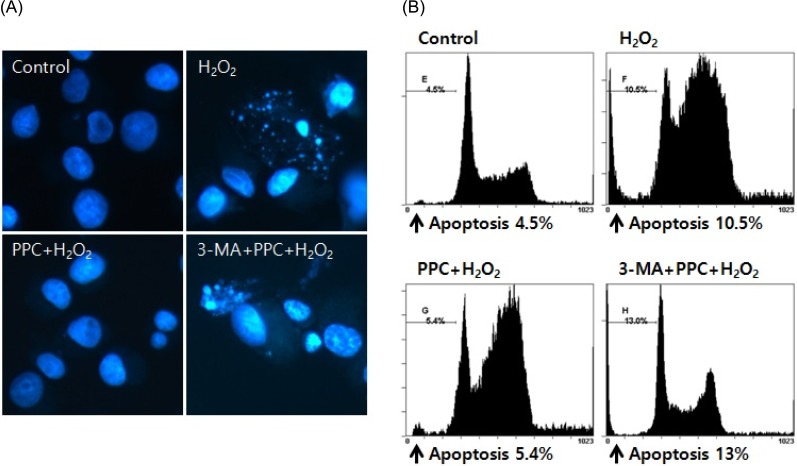
2. Propofol attenuated H2O2-induced COS-7 cell apoptosis
The effect of propofol on COS-7 cell apoptosis was examined using Hoechst 33342 staining. The cells were viewed under a fluorescence microscope (× 400) to evaluate the morphological changes in the nuclei (Fig. 3A). The majority of COS-7 cells in the control group showed normal morphology with round regular nuclei. In contrast, apoptotic nuclei, characterized by bright blue chromatin that was highly condensed or fragmented, were observed in the H2O2 and 3-MA + PPC + H2O2 group cells. Pretreatment with propofol effectively decreased COS-7 cell apoptosis, as indicated by the restored cell morphology. Upon determining apoptosis by using a fluorescence-activated cell sorter, we found that 10.5% of the cells showed apoptosis in the H2O2 group, but in the PPC + H2O2 group, only 5.4% of the cells showed H2O2-induced apoptosis. These results suggested that PPC attenuated H2O2-induced COS-7 cell apoptosis. However, pretreatment with 3-MA inhibited the protective effect of propofol against apoptosis, and the percentage of apoptotic cells increased to 13% in the 3-MA + PPC + H2O2 group compared to the PPC + H2O2 group (Fig. 3B).
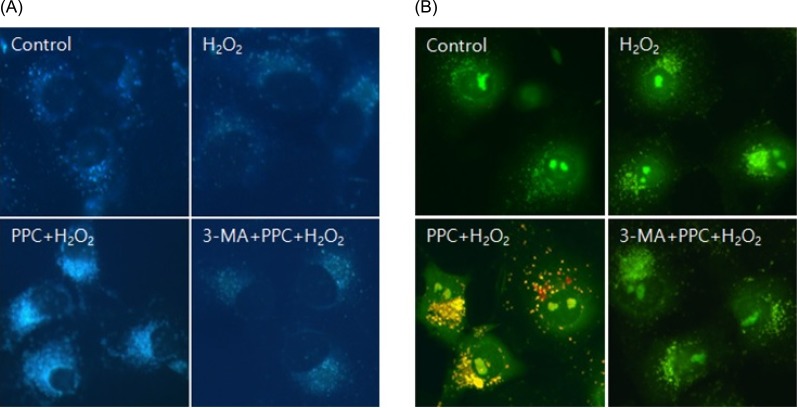
3. Propofol resulted in the induction of autophagy in COS-7 cells
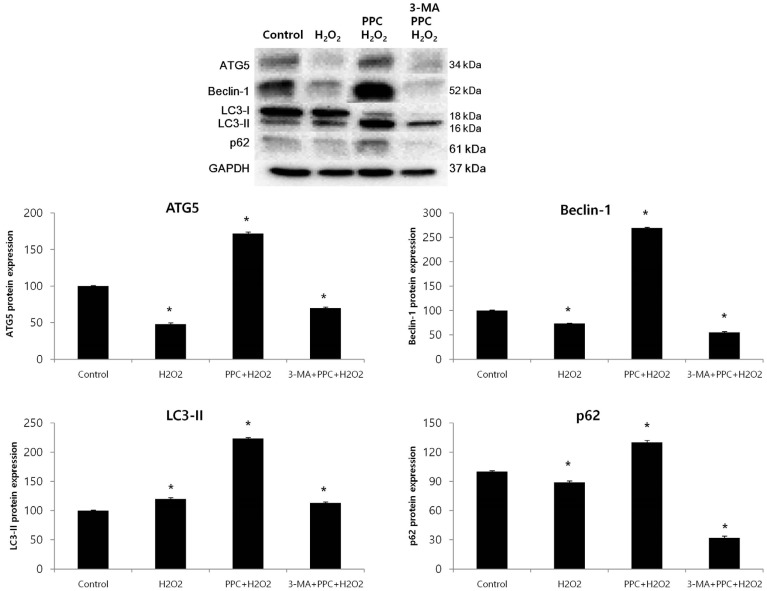
Prominent accumulation of the autophagy-specific stain MDC was observed around the nuclei in the PPC group COS-7 cells (Fig. 4A). We clarified the role of propofol-induced autophagy in COS-7 cells by investigating the consequences of treatment with 3-MA, a selective autophagy inhibitor. The inhibitory effects of 3-MA on AVO formation was confirmed by quantitatively measuring the red fluorescence ratio after AO staining. Yellow and red fluorescent spots appeared in the propofol-pretreated COS-7 cells, while the cells in the control and 3-MA + PPC + H2O2 groups showed mainly green cytoplasmic fluorescence (Fig. 4B). We examined the activation of autophagy-related proteins in the H2O2-treated cells by using western blot analysis. The levels of Atg5, Beclin-1, and p62 decreased more significantly in the H2O2 group than in the control group after the H2O2-induced apoptotic signaling. The levels of Atg5, Beclin-1, and p62 increased when autophagy was induced by propofol, and their levels decreased when autophagy was suppressed by 3-MA. The level of LC3-II increased more significantly in the H2O2 group than in the control group and increased even more in the PPC + H2O2 group. However, pretreatment with 3-MA inhibited LC3-II expression (Fig. 5).
DISCUSSION
In this study, the protective effect of PPC against oxidative-stress-induced COS-7 cell apoptosis was proven in an in vitro experimental model of H2O2-induced oxidative injury. We also demonstrated that the antioxidant effect of PPC was mediated by cellular autophagy activation by showing the formation of autophagosomes in COS-7 cells by using a fluorescence microscope and the expression of autophagy-related proteins by using western blot analysis.
Oxidative stress can induce toxic effects through the production of ROS that damage cardinal cellular components, such as proteins, lipids, and DNA, because of their highly reactive nature [20]. Navarro et al. [21] reported that H2O2 was associated with cellular toxic effects, including the depletion of intracellular glutathione and ATP, an increase in NAD+ level, increase in free cytosolic Ca2+, and lipid peroxidation. Among the ROS, the hydroxyl radical can directly attack the DNA backbone by generating oxidative damages, such as oxidized bases, abasic sites, single-strand breaks, double-strand breaks, and DNA-protein crosslinks [2223]. Via these processes, oxidative stress is involved in the development of cardiovascular, neurological, and ophthalmological diseases and cancer, as well as in aging [1]. Therefore, we need to develop strategies for preventing oxidative injury.
The mediators associated with the cellular signaling pathway that respond to oxidative injury are the extracellular signal-regulated kinase (ERK), c-Jun amino-terminal kinase (JNK), and p38 MAPK signaling cascades; the phosphoinositide 3-kinase (PI3K)/Akt pathway; the nuclear factor (NF)-kB signaling system; p53 activation; and the heat shock response. In general, the ERK, PI3K/Akt, and NF-kB signaling pathways and the heat shock response produce a pro-survival effect during oxidative injury, whereas the activation of JNK, p38 MAPK, and p53 is more commonly involved in apoptosis [1]. Propofol exerts a protective effect against oxidative injury via these mediators. Wang et al. [24] demonstrated that propofol protects hepatocytes from H2O2-induced apoptosis, partly through the activation of the ERK pathway. In another study, they found that propofol suppressed p38 MAPK activation and attenuated oxidative-stress-induced apoptosis [25]. Previous reports have shown that heme oxygenase (HO) plays a critical role in the antioxidant pathway of propofol [262728] and Acquaviva et al. [29] reported that the addition of a synthetic NF-kB inhibitor completely reversed propofol-mediated HO-1 expression, suggesting that propofol has a protective effect on oxidative injury via NF-kB activation.
In our study, COS-7 cells were pretreated with the autophagy-pathway inhibitor 3-MA and propofol before they were exposed to H2O2-induced oxidative stress, and we investigated the changes in COS-7 cell apoptosis based on the assumption that autophagy might play an important role in the protective effect of propofol against oxidative stress. Hoechst 33342 staining revealed that the number of apoptotic bodies was higher in the 3-MA + PPC + H2O2 group cells than in the control and PPC + H2O2 group cells. This result suggests that autophagy has a cytoprotective function related to the protective mechanisms of PPC against oxidative injury. A recent study demonstrated that oxidative stress induced autophagy through molecular crosstalk between p62, an autophagy-related protein, and transcription factors related to cytoprotective genes in response to oxidative stress, and these had a protective effect against necrosis due to ATP depletion [30]. Other studies have shown that the suppression of autophagy by knockdown of Atg5 and Atg7, which are autophagy-related genes, greatly increased H2O2-induced apoptosis; these studies also showed that autophagy played a cytoprotective role in H2O2-induced apoptosis [3132]. In this study, PPC upregulated the expression of autophagy-related proteins, such as Atg5, Beclin-1, and p62.
The molecular circuitry and signaling pathways regulating autophagy involve five key stages: (a) phagophore formation or nucleation; (b) Atg5-Atg12 conjugation, interaction with Atg16L, and multimerization at the phagophore; (c) LC3 processing and insertion into the extending phagophore membrane; (d) capture of random or selective targets for degradation; and (e) fusion of the autophagosome with the lysosome, followed by the proteolytic degradation of engulfed molecules by lysosomal proteases [33]. LC3 is expressed upon the induction of autophagy and is converted to LC3-II by LC3 processing. LC3 usually reveals two bands: LC3-I (18 kDa) and LC3-II (16 kDa). The amount of LC3-II is related to the number of autophagosomes and is used for analyzing autophagic activity [34]. The recruitment and integration of LC3-II into the growing phagophore, including LC3 processing, are dependent on Atg5-Atg12. The Atg5-Atg12–dependent pathway of autophagy has been considered critical for survival during the starvation period in the first few days after birth [3536]. However, a recent study discovered that the Atg5-Atg12–independent pathway was an alternative process for autophagy. This pathway of autophagy was not associated with LC3 processing but appeared to specifically involve autophagosome formation from trans-Golgi and late endosomes [37]. In our study, the expressions of Atg5, Beclin-1, and p62 decreased as a result of H2O2-induced oxidative stress, but LC3-II expression was higher in the treatment groups than in the control group. This finding suggests that LC3 processing is generated by an Atg5-independent pathway during H2O2-induced oxidative stress and that PPC activates the production of Atg5, thereby resulting in the increase of LC3-II expression through the Atg5-independent and Atg5-dependent pathways in the PPC + H2O2 group. After all, the levels of autophagy-related proteins were higher in the PPC + H2O2 group than in the control group, suggesting that PPC enhanced autophagy. However, we did not investigate the roles of PI3K/Akt, p38 MAPK, and JNK, which are known to be involved in the autophagy signaling pathway. This is a limitation of our study.
In conclusion, this study suggests that PPC has a protective effect on H2O2-induced COS-7 cell apoptosis and this effect seemed to be mediated by autophagy. Whether autophagy has a positive influence on oxidative injury and cell survival remains a controversial topic, and more research is needed to clarify the role of autophagy. Nevertheless, our study provides evidence that propofol inhibits oxidative-stress-induced apoptosis through the induction of cellular protective autophagy.
 XML Download
XML Download