

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Human skin is composed of tightly connected keratinocytes in the stratum granulosum and terminally differentiated keratinocytes in the stratum corneum. This intricate constitution comprises an effective barrier that resists extensive water loss while simultaneously protecting the cell from the entry of microbial pathogens and allergens [1]. Surgical damage can disrupt this barrier, and a fair number of patients have wound healing abnormalities [2]. In addition, non-healing wounds can deteriorate chronic ulcer or cause wound-related secondary infections, and these conditions make it harder to recover from disease. At worst, chronic wound tissue can develop into cancer due to persistent inflammation [3].
Propofol (2,6-diisopropylphenol) is a widely used agent for the induction and maintenance of anesthesia during surgical procedures. It is also used as a sedative for intensive care unit (ICU) patients. Propofol has structural similarity to the endogenous antioxidant vitamin E and shows antioxidant activity [4]. Propofol has a protective effect on hydrogen peroxide (H2O2)-induced apoptosis in cardiac cells [5] and in myocardial ischemia and reperfusion injury in rats [6]. However, the effect of propofol on human keratinocytes under oxidative stress is yet to be fully elucidated. In this study, we investigated how propofol influences intracellular autophagy and apoptotic cell death induced by oxidative stress in human keratinocytes.
MATERIALS AND METHODS
1. Reagents
Propofol (Fresenius Kabi Austria Gmbh, Hafnerstrasse, Austria) diluted with dimethyl sulfoxide (DMSO), Hoechst 33342, and chloroquine were purchased from Sigma (St. Louis, MO, USA). The following reagents were obtained commercially: 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT), acridine orange (AO), monodansylcadaverine (MDC), and 3-methyladenine (3-MA, class III PI3K inhibitor) (Calbiochem, La Jolla, CA, USA). The antibodies used in the study were as follows: Bak (1:1000), caspase-9 (1:1000), caspase-3 (1:1000), and poly (ADP-ribose) polymerase (PARP; 1:1,000) (Abcam, Cambridge, MA, USA).
2. Cell culture
Human keratinocytes (HaCaT cell line) were obtained from the American Type Culture Collection (ATCC, Manassas, USA). Dulbecco's modified Eagle's medium (DMEM; GIBCO, Life Technologies, Carlsbad, CA, USA) supplemented with 10% inactivated fetal bovine serum (FBS, GIBCO) containing 500 µg/ml penicillin and 500 µg/ml streptomycin (GIBCO) was used to culture the cells at 37℃ in a humidified environment with 5% CO2. The medium was changed once every 3 days.
3. Oxidative stress of cultured human keratinocytes and drug treatment
The propofol solution was prepared by dissolving propofol in DMSO, which was kept frozen at −4℃ until use. The stock was diluted to the appropriate concentration with DMEM when needed. Prior to propofol treatment, cells were grown to approximately 75% confluence and then exposed to propofol at various concentrations (0, 3, 30, 100, and 300 µM) for 2 h. The following four groups were used for experimentation: control, cells were incubated at 37℃ in a humidified atmosphere with 5% CO2 without propofol treatment;. H2O2, cells were exposed to 300 µM hydrogen peroxide (H2O2) for 2 h; PPC/H2O2, cells were pretreated with 100 µM propofol for 2 h and exposed to 300 µM H2O2 for 2 h; and 3-MA/PPC/H2O2, cells were pretreated with 1 mM 3-MA and 100 µM propofol for 1 h and 2 h, respectively. After treatment of 3-MA and propofol, cells were exposed to 300 µM H2O2 for 2 h (Fig. 1).
4. Cell viability assay
The cell viability was determined using an MTT assay. Cells were cultured in 96-well plates (4×103 cells/well). The cells were then treated with the indicated concentrations of drug. At the end point of treatment, 100 µl of MTT (500 mg/ml) was added to each well. The cells were incubated for 4 h at 37℃. Then the formazan crystals were solubilized in DMSO (200 µl/well) by constant shaking for 15 min. The cell viability was measured using an ELISA reader (Tecan, Männedorf, Switzerland) at a 620 nm excitatory emission wavelength.
5. Hoechst staining
Cells were harvested and transferred to a clean, glass slide using a cytocentrifuge. Cells were stained with 1 µg/ml Hoechst 33342 for 15 min at 37℃ in the dark and washed with phosphate buffered saline (PBS). The slides were mounted with glycerol. The samples were observed and photographed under an epifluorescence microscope (Carl Zeiss, Goettingen, Germany).
6. Measurement of mitochondrial membrane potential (MMP)
DiOC6 (Molecular Probes, Eugene, OR, USA) was added to the medium (1 µM final concentration) and incubated for 30 min. To investigate MMP, flow cytometry was performed using a CYTOMICS FC500 system (Beckman Coulter, Brea, CA, USA). The data were analyzed using CXP software version 2.2. The analyzer's threshold on the FSC channel was adjusted to exclude noise and most of the subcellular debris.
7. MDC and AO staining
After chemical treatment of the four groups of HaCaT cells, cells were stained with 1.25 µM MDC, a selective fluorescent marker for autophagic vacuoles, at 37℃ for 30 min. The cellular fluorescence changes were observed using a fluorescence microscope (Axioskop, Carl Zeiss, Göettingen, Germany). As an autophagy control, cells were starved using EBSS. For further detection of the acidic cellular compartment, AO was used, which emits bright red fluorescence in acidic vesicles, but fluoresces green in the cytoplasm and nucleus. The cells were stained with 1 µg/ml AO for 5 min and washed with PBS. The formation of acidic vesicular organelles (AVOs) was observed using an LSM 700 confocal microscope (Carl Zeiss).
8. Western blot analysis
The cells (1.5×106) were washed twice in ice-cold PBS, resuspended in 200 µl of ice-cold solubilizing buffer (300 mM NaCl, 50 mM Tris-Cl (pH 7.6), 0.5% TritonX-100, 2 mM PMSF, 2 µl/ml aprotinin, and 2 µl/ml leupeptin) and incubated at 4℃ for 1 h. The lysates were centrifuged at 13,200 rpm for 30 min at 4℃. Protein concentrations of cell lysates were determined using a Bradford protein assay (Bio-Rad, Richmond, CA, USA), and 20 µg of proteins were resolved by 10% SDS/PAGE gel. The gel was transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking, the membranes were reacted with the appropriate primary antibodies. Immunostaining with secondary antibodies was detected using SuperSignal West Femto substrate (Pierce, Rockford, IL, USA).
9. In vitro wound healing
Cells were seeded into a cover glass for microscope slides in DMEM with 10% FBS. The confluent cell basal layer was scratched with an Eppendorf sterile pipette tip to steadily produce a circular cell-free zone (1 mm in diameter) on the coverslips. The wound cultures were then incubated at 37℃ for 24 h, and the wound healing process was observed under optical microscopy. After 24h of incubation in growth medium at 37℃, the new growth and migration into the cell-free zone was considered as the process of in vitro healing. Images of the relatively healed monolayer area were obtained and the pixel counts were analyzed using NIH ImageJ software.
RESULTS
1. Propofol improved cell viability under oxidative stress
The effect of propofol was investigated over a wide concentration range. Propofol preconditioning and 3-MA treatment had no influence on cell viability in the normoxic state. However, a concentration of more than 300 µM H2O2 decreased cell viability (Fig. 2A-C). Based on this result, all subsequent experiments were performed with 100 µM propofol and 300 µM H2O2. The cell viability rate (%) was calculated and compared to the control group. The H2O2 and 3-MA/PPC/H2O2 groups had lower cell viabilities than the control group did (Fig. 3).
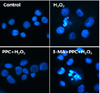
2. Propofol protected against oxidative stress-induced apoptosis in human keratinocytes
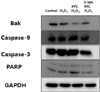
Hoechst 33258 staining was used for detection of apoptosis under a fluorescence microscope (× 400) (Fig. 4). The majority of cells in the control and PPC/H2O2 groups exhibited round regular nuclei considered as normal morphology. In contrast, apoptotic bodies were seen in cells of the H2O2 and 3-MA/PPC/H2O2 groups. Moreover, flow cytometry was used to detect the percentage of apoptotic cells (Fig. 5). Compared to that in the control group and PPC/H2O2 group, the number of apoptotic cells increased in the H2O2 and 3-MA/PPC/H2O2 groups. To ascertain the effect of propofol on apoptosis, we also examined the expression of factors associated with apoptosis in the cells subjected to oxidative stress by western blot analysis. In the PPC/H2O2 group, cellular expression of caspase-9, caspase-3, and PARP increased while expression of Bak was down-regulated (Fig. 6).
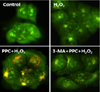
3. Propofol led to the activation of autophagy in human keratinocytes
A significant accumulation of autophagy-specific staining of MDC was observed around the nuclei in cells of the PPC/H2O2 group (Fig. 7). Similar results were observed upon AO staining; red fluorescent spots were observed in the PPC/H2O2 group cells, while the control, H2O2, and 3-MA/PPC/H2O2 groups showed mainly green cytoplasmic fluorescence (Fig. 8).
4. Propofol stimulated cell migration in the wound-healing assay
An in vitro wound-healing assay was performed to determine the migratory capability of human keratinocytes. Wound healing was monitored at 0 and 24 h. The PPC/H2O2 group treated with propofol showed increased wound healing capability (Fig. 9).
DISCUSSION
Skin cells are equipped with an elaborate system that maintains the balance between oxidative stress and antioxidant defense. This system can protect the cells from oxidative stress damage [7]. The excessive cellular levels of reactive oxygen species, such as singlet oxygen, superoxide anions, and H2O2 have been linked to apoptosis. This oxidative stress can induce the damage of cellular proteins, lipids, and polysaccharides, and may also indirectly cause structural damage to DNA [8910].
The biology of keratinocytes is divided into two pathways associated with cell death: apoptosis and differentiation. The pathway that leads to terminal differentiation is known as cornification, which has several steps. Other authors have described this process as a specialized form of programmed cell death, in which the cells are not removed by phagocytosis like the apoptotic process, and it is used to maintain homeostasis in the skin [11].
Apoptosis is a complex process of purposeful suicide by a cell in a multi-cellular organism. It is one of the main types of programmed cell death and involves biochemical events leading to a characteristic cell morphology and death [12]. Autophagy, an evolutionary conserved process of “self-eating,” occurs at a basal level in most cells and contributes to the turnover of long-lived proteins and organelles to maintain intracellular homeostasis [13]. Accordingly, apoptosis and autophagy constitute the two main pathways through which superfluous, damaged, or aged cells and organelles are eliminated.
Previous reports have shown that the cellular protective mechanism of propofol may partly result from its ability to act as a free radical scavenger [141516]. The aim of this study was to determine the protective effect of propofol on human keratinocytes against oxidative stress and to investigate whether autophagy is associated with this protective mechanism. From this study, we present four principal findings.
First, propofol preconditioning before oxidative stress increased the proliferation of human keratinocytes under oxidative stress conditions via MTT assay, and 3-MA treatment blocked this protective effect of propofol preconditioning.
Second, propofol preconditioning exerted an antiapoptotic effect on human keratinocytes exposed to oxidative stress. In western blot analysis, we found that propofol preconditioning increased the cellular expression of caspase-9, caspase-3, and PARP, and decreased Bak expression. Among the factors associated with apoptosis, caspase-9, caspase-3, and PARP play an important role in the caspase-dependent pathway, and the pro-apoptotic protein Bak is related to the mitochondria-dependent apoptotic pathway [171819].
Third, the anti-apoptotic effect of propofol preconditioning in human keratinocytes was probably due to the induction of intracellular autophagy. 3-MA is an intracellular autophagic pathway inhibitor that increases cellular apoptosis, and is suggested to play an important role in intracellular autophagy in the anti-apoptotic effect of propofol preconditioning. In the autophagy-specific staining with MDC and AO, the PPC/H2O2 group showed greater expression of the autophagosome than the other groups did.
Finally, an in vitro wound-healing assay was performed to determine the migratory ability of human keratinocytes. The PPC/H2O2 group showed increased migratory capability. Migration of keratinocytes is an essential process in wound healing [20].
Based on these results, although the current study could not demonstrate the protection by propofol preconditioning in vivo, we suggest that propofol preconditioning induced an endogenous cellular protective effect in human keratinocytes against oxidative stress through the activation of signaling pathways related to autophagy. Finally, we believe that our results would contribute to the body of knowledge regarding the relationship between wound healing and oxidative stress.






 XML Download
XML Download