

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The increasing prevalence of Alzheimer disease (AD) is related to the extended lifespan of human-being. As social and medical costs for the treatment of AD has become a significant burden, there has been the increased attention on the effective preventive strategies against AD.
Excessive accumulation of amyloid beta (Aβ) peptide and tau protein on the brain tissue is a well-known etiology of AD [1]. Therefore, Aβ peptide and tau protein are often considered as potential pharmacological targets. Amyloid precursor protein (APP) is cleaved by β-secretase 1, a beta-site APP cleaving enzyme 1, to produce Aβ peptide [2]. Presenilin 1 (PS1) is a part of γ-secretase complex which cleaves APP [3]. Therefore, the presence of APP and PS1 induces increased Aβ peptide production in brain region. Excessive soluble and aggregated Aβ peptides and hyper-phosphorylation of tau protein stimulate plaque formation and structural/functional alterations of neurons in brain [4]. These deleterious alterations are reported to be accompanied by increased oxidative stress as well as pro-inflammatory response [56].
Niacin is a vitamin B3 involved in energy metabolism, mitochondrial function, cell death, and aging as a form of nicotinamide adenine dinucleotide (NAD+). Nicotinamide (NAM) treatment suppressed poly (ADP-ribose) polymerase-1 over-activation in a rat model of AD [7]. Nicotinamide riboside (NR), 1 of 3 different forms of niacin, is a NAD+ donor, and improves cognitive function with the alterations of proliferator-activated receptor-γ coactivator 1α, β-secretase 1, and mitochondrial genes in AD mouse models [89]. These studies intended to investigate the treatment effects of niacin, however, the preventive effect of niacin against AD has not been reported.
Therefore, the purpose of this study is to investigate whether 2 types of niacin, nicotinic acid (NA) and NAM reduce the AD-related genes in brain tissues of Aβ-injected mice.
MATERIALS AND METHODS
Animals and treatments
Male Crj:CD1 (ICR) mice at 9 weeks of age were housed in individual cages at controlled temperature (23°C ± 3°C) and relative humidity (50% ± 5%) with a 12 hours light/dark cycle. The mice were fed standard irradiated rodent chow (10% kcal from fat; Research Diets, New Brunswick, NJ, USA) with unlimited access to food and water for a 1-week acclimation period. Then, the mice were divided into 6 groups (n = 5 mice per group), and fed with the assigned diets (standard chow diet; Research Diets) and treatments. The experimental groups were as follows: 1) control, 2) Aβ control (Aβ-CON), 3) Aβ + NA (Sigma-Aldrich, St. Louis, MO, USA) 20 mg/kg/day (NA20), 4) Aβ + NA40, 5) Aβ + NAM (Sigma-Aldrich) 200 mg/kg/day (NAM200), and 6) Aβ + NAM400. After 1-week acclimation period, the mice orally received vehicle (phosphate buffered saline [PBS]), NA, or NAM once a day for a total of 7 successive days. On day 7, either 100 µL of vehicle (PBS) or biotinylated Aβ42 (100 µM in PBS; Abcam, Cambridge, UK) was injected into mouse tail vein. This study protocol conformed to the specifications outlined in the National Institutes of Health Guiding Principles for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee of Chonnam National University (approved protocol No. CNU IACUC-YB-2015-4).
Blood and tissue collection
At 5 hours after the injection of vehicle or biotinylated Aβ42 injection, fasted mice were anesthetized with an intraperitoneal injection of each 0.1 mL mixture of Zoletil (10 mg/kg) and Rompun (5 mg/kg) at the ratio 2:1 in the mouse. Blood was collected by cardiac puncture. Tissues were harvested, weighted, and stored at −80°C until further analysis.
Western blot analysis
Total protein was isolated from tissues by homogenization in cold Radio-Immunoprecipitation Assay (RIPA) lysis buffer (Amresco, Solon, OH, USA) containing protease inhibitors and phosphatase inhibitors (Sigma-Aldrich). The lysates were centrifuged, and supernatants were collected and subjected to western blot analysis. Protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer's instructions. Western blotting was performed by denaturing 50 µg of protein at 95°C for 5 minutes in Laemmli sample buffer (Fermentas, Burlington, Canada). Sample proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane. Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline/Tween-20 (50 mM Tris, pH 7.5, 500 mM sodium chloride, and 0.05% Tween-20) for 1 hour at room temperature. Membranes were incubated overnight at 4°C with primary antibodies for Aβ (Abcam). Membranes were then exposed to an anti-rabbit secondary antibody conjugated to horseradish peroxidase (Cell Signaling Technology, Danvers, MA, USA) for 1 hour at room temperature. Signals were detected by chemiluminescence using the enhanced chemiluminescence (ECL) detection reagent (GE Healthcare, Piscataway, NJ, USA). The bands were scanned by a Geliance 600 Imaging System (PerkinElmer, Waltham, MA, USA) with a cooled 12-bit camera, and quantified by densitometry.
Isolation of total RNA and quantitative reverse transcription-polymerase chain reaction (PCR)
Total RNA was isolated from tissues with a PureLink RNA Mini kit (Invitrogen, Carlsbad, CA, USA). Reverse transcription was performed using a SuperScript III First-Strand Synthesis System (Applied Biosystems, Foster city, CA, USA) following the manufacturer's instructions. mRNA expression was quantified by real-time PCR (StepOnePlus; Life Technologies, Carlsbad, CA, USA). Synthesized cDNA was mixed with Power SYBR Green PCR Master Mix (Applied Biosystems) and a gene-specific primer (Bioneer, Daejeon, Korea). Individual reactions for target and 18S were carried out separately with negative controls lacking cDNA. The conditions used were as follows: 95°C for 10 minutes, followed by 40 cycles of denaturation (95°C for 10 seconds), annealing (Tm [°C] for 15 seconds), and extension (72°C for 60 seconds). The cycle number for threshold of detection was determined by StepOne Software (Life Technologies). mRNA expression of each target was normalized to that of 18S gene and expressed as fold change relative to controls.
Statistical analysis
All statistical analyses were performed using SPSS Statistics 23 (SPSS Inc., Chicago, IL, USA). Data are expressed as means ± standard error of the mean (SEM). Student's t-test or one-way analysis of variance (ANOVA) as well as post-hoc analysis was performed to compare the differences among groups. Statistical significance was defined as p < 0.05.
RESULTS
Aβ42 tail vein injection increased brain Aβ42 levels
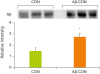
After 7-day pre-treatment of vehicle, NA or NAM, vehicle, or Aβ was injected via tail vein. Body weight, food intake, and tissue weights were not affected by the NA or NAM treatments and/or Aβ42 tail vein injection (data not shown). Western blot analysis confirmed that Aβ42 tail vein injection significantly increased Aβ42 protein in brain tissue (Figure 1).
NAM400 pre-treatment reduced APP, PS1, and nuclear factor kappa B (NF-κB) in brain tissues
Aβ42 injection tended to increase brain APP expression (p = 0.061), and significantly decreased brain sirtuin 1 (Sirt1) expression (Figure 2A and 2C). NA40 increased brain APP expression compared with Aβ-CON, which suggests that NA40 may be a high dose to be able to exert deleterious effects irrespectively of the up-regulation of Sirt1 by NA40 treatment.
Figure 2
NAM reduces 2 AD-related targets (A) APP and (B) PS1, and inflammation-related targets (C) Sirt1 and NF-κB in brain tissues of Aβ tail vein-injected mice. Data are expressed as mean ± SEM. Different letters within a variable are significantly different at p < 0.05.
NAM, nicotinamide; AD, Alzheimer disease; APP, amyloid precursor protein; PS1, presenilin 1; Sirt1, sirtuin 1; NF-κB, nuclear factor kappa B; SEM, standard error of the mean; CON, control; NA, nicotinic acid.

NA200 pre-treatment had no effect on the expression of APP and PS1. NAM400 pre-treatment significantly reduced gene expression of APP and PS1 in brain tissues (Figure 2A and 2B). And, NAM200 and NAM400 pre-treatments significantly increased Sirt1 expression in brain tissues, which is accompanied by the decreased brain expression of NF-κB by 2 doses of NAM (Figure 2C and 2D).
DISCUSSION
In the present study, we found that Aβ42 tail vein injection increased Aβ42 deposition in brain tissue, and demonstrated that NAM treatments down-regulated gene expression of AD-related genes, APP and PS1 in brain tissues of Aβ42-injected mice. The NAM-mediated alterations of 2 AD-related genes were accompanied by the up-regulation of Sirt1 and the down-regulation of NF-κB in the brain tissues.
Aβ is produced by neural and non-neural cells, and circulated in blood and cerebrospinal fluid. Aβ is cleared out to be a well-balanced status between synthesis rate and clearance rate in a normal condition [10]. As the body gets aged or injured by environmental and pharmacological factors, Aβ deposition is increased in the regions of extracellular fluid and cerebral vessels, which subsequently impairs neural integrity and functions. In rodent models of AD, up-regulation of APP and PS1 is one of the ways to increase Aβ synthesis, and hyper-phosphorylation of tau protein stimulates inflammatory responses in the affected regions [24]. In this study, NAM pre-treatment down-regulated APP and PS1 in Aβ-injected mouse brains, which suggests that NAM may have preventive effects against the Aβ deposition and Aβ-mediated alterations in neuron structure and function.
Recent papers suggested the amyloid cascade-inflammatory hypothesis that Aβ deposit leads to pro-inflammatory responses in neuronal cells, especially microglia [6]. This hypothesis implies the possibility that AD is related to other inflammation-related metabolic diseases and that anti-inflammatory drugs or nutrients may prevent or delay the development and progression of AD. Niacin exerts anti-inflammatory effects in a rodent model of cognitive impairment, diabetes, and non-alcoholic fatty liver diseases [1112]. NAM was protective against sevoflurane-induced cognitive impairment in rats, and suppressed the activation of NF-κB and caspase-3 [11]. NR reduced hepatic pro-inflammatory markers (tumor necrosis factor-alpha, interleukin-6, and interleukin-1) and nucleotide binding and oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome components (NLRP3, adaptor proteins like apoptosis-associated speck-like protein containing a caspase activation and recruitment domain, and caspase1) in obese and diabetic mice [12]. Niacin increases the activity of Sirt as a NAD+ donor [13]. Sirt is involved in glucose and lipid metabolism as well as inflammation [1415]. Activation of Sirt exerts anti-inflammatory effects in a rodent model [14]. Here, in addition to the modulating effect of NAM's on 2 AD-related genes, APP and PS1, NAM altered the gene expression of Sirt1 and NF-κB in Aβ-injected mouse brains, suggesting that there may be a link between Aβ deposit and inflammatory response.
Although this report has a main limitation in lacking of in-depth mechanistic investigation, these preliminary and interesting findings suggest that NAM supplementation may be a potential preventive strategy against AD-related deleterious change, and further mechanistic studies are also needed.
 XML Download
XML Download