

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ependymoma is a central nervous system (CNS) tumor that accounts for 3% to 6% of all CNS tumors [1]. They are glial tumors that usually arise from the ependymal cells lining the ventricles and central canal within the spinal cord. The distribution of these tumors along the neuro-axis varies by age and most commonly involves the spinal cord in adults and the posterior fossa in children [2]. They may also occur outside the ventricular structure as rare ectopic ependymomas. Pituitary ependymomas are extremely rare and have been reported only seven times in the literature [1345678910]. Except one case from Belcher et al. [11], who reported a case with a 28-year follow-up, no long-term follow-up results have been reported. We present a patient with a pituitary ependymoma who underwent partial tumor resection via the trans-sphenoidal approach with postoperative radiation therapy 10 years ago. Follow-up magnetic resonance imaging (MRI) after 10 years of treatment shows no evidence of recurrence.
CASE REPORT
Clinical presentation
A 59-year-old male patient presented with fatigue, general weakness, erectile dysfunction, and loss of body hair, including pubic hair. The visual acuity showed 0.2 in the right eye and 0.8 in the left eye, and the visual field examination revealed bitemporal hemianopsia. There was no other neurological deficit except visual disturbance. The lab findings showed changes compatible with panhypopituiarism with low levels of thyroxine, cortisol, and testosterone. Complete blood count was hemoglobin 13.4 g/dL (13.0–17.0), white blood cells 7.28×103/µL (4.0–10.0), and platelet 278×103/µL (130–400). Serum electrolyte was sodium 140 mmol/L (135–145), potassium 3.0 mmol/L (3.5–5.5), chloride 106 mmol/L (98–110). Serum free T4 was 0.39 ng/dL (0.89–1.79), serum thyroid-stimulating hormone 3.90 µIU/mL (0.17–4.05), serum cortisol less than 1.0 µg/dL (4.3–22.4), plasma adrenocorticotropic hormone 18.3 pg/mL (6–60), and serum testosterone less than 0.01 ng/mL (1.81–7.58). The thyroid glands showed diffuse volume loss without focal lesion, suggesting hypothyroidism on thyroid ultrasonography.
Imaging findings
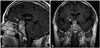
Initial sellar MRI revealed a 3.3×3.5×2.3-cm sellar and suprasellar snowman-shaped enhancing mass (Fig. 1). Preoperative radiologic diagnosis was a pituitary macroadenoma. The lesion was homogeneous, solid, and hypo-enhanced compared with normal pituitary gland. It compressed the overlying optic chiasm, and there was no definite evidence of cavernous sinus invasion. According to the reading of MRI and a basal pituitary hormone study, it was compatible with non-functioning pituitary adenoma.
Operation
The tumor was partially resected via the trans-sphenoidal approach. There was no discernible surgical findings suspecting ependymoma. At first, we couldn't assume that the tumor was ependymoma. However, the mass was firm and bloody, unlike usual pituitary adenoma. There were no postoperative complications.
Histopathological findings
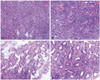
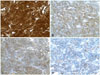
Microscopically, the tumor was highly cellular and composed of compactly arranged small round to oval cells. In area, the tumor showed glandular or papillary structures. These features were very similar to those of pituitary adenoma at a glance. In higher magnification, tumor cells showed somewhat elongated nuclei, and arranged in the fibrillary background, forming perivascular pseudorosettes (Fig. 2A and B), papillary configurations (Fig. 2C), or true ependymal rosettes (Fig. 2D). Immunohistochemical stains showed strong positive reactions to vimentin and S-100 protein (Fig. 2A). The tumor was also positive for S100 protein and glial fibrillary acidic protein (Fig. 3A and B) and CD99 (Fig. 3C). In epithelial membrane antigen staining, characteristic paranuclear dot-like positivity was also noted (Fig. 3D). The histologic and immunohistochemical findings were compatible with ependymoma. Mitoses were not found and there was no necrosis. Immunohistochemical stain for Ki-67 showed the labeling index was less than 1%.
Postoperative course
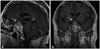
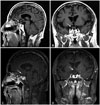
Immediate postoperative MRI showed a 3.0×2.3-cm residual tumor in the suprasellar and sellar space (Fig. 4). It showed reduction of mass effect upon the optic chiasm. The patient was histopathologically diagnosed with an intrasellar ependymoma. After 2 months, he received adjuvant radiation therapy of 54 Gy over 30 fractions to intrasellar lesion for 6 weeks. A followed-up MRI was taken 3 months after radiation therapy, and it showed markedly decreased size of mass (Fig. 5A and B). It decreased to 1.7×2.1-cm sized mass, and its compressive effect on optic chiasm was much relieved.
Thyroid and steroid hormones were replaced. Symptoms of fatigue, general weakness, and visual field defect improved. There was no newly developed neurological deficit. Follow-up MRIs were performed every 3 months initially, then every 6 months. At the 10-year follow up, MRI revealed no evidence of tumor progression (Fig. 5C and D).
DISCUSSION
Ependymomas usually arise from ependymal cells, lining ventricles or central canals. Because pituitary lesion has no anatomical relevance to ependymal cell lining, pituitary ependymomas are extremely rare. One theoretical explanation for pathogenesis of pituitary ependymoma is that a dysembryogenic component of faulted migration pattern could be in association with inadequate apoptotic potential [9].
A literature review showed that eight cases, including the current case, have been reported. All previously reported cases were diagnosed by pathologic confirmation after surgical resection. Belcher et al. [11] presented a patient with pituitary ependymoma who underwent four operations, adjuvant radiation therapy, and chemotherapy within 28 years of follow-up. The other six cases were followed up for less than one year, and the masses were only surgically removed without adjuvant therapy [345678910]. We present a patient having partial removal of tumor followed by adjuvant radiotherapy with 10 years of follow-up without tumor progression.
There is no established diagnostic and treatment protocol for pituitary ependymoma; however, surgical resection should be performed in advance for histopathological diagnosis. Although there is no definite differential diagnostic point between pituitary adenoma and ependymoma in clinical presentations and preoperative examinations, it could be helpful in that enhancement of adenoma is slower than that of normal gland in contrast-enhanced MRI. On the other hand, ependymoma is simultaneously enhanced with normal pituitary gland.
Although the histopathologic evaluation of ependymoma is usually benign, its 5-year survival rate is usually lower than that of meulloblastoma due to its relative lack of sensitivity to radiation therapy and chemotherapy in most cases. Pituitary ependymoma is extremely rare with lack of knowledge about diagnosis and treatment of the tumor. Our case is the only case in which the disease has been well controlled over long period without tumor progression, and it could suggest the most effective method to treat the disease. If the tumors are decompressed and distance is left from the optic apparatus and carotid, ependymomas can be controlled by partial removal with high doses of postoperative local radiotherapy. The therapeutic method in our case could be the most effect way to treat pituitary ependymoma.
 XML Download
XML Download