

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ependymomas is a tumor that arises from the ependyma which is more common in pediatric group and tends to occur intracranially. Common location of intracranial ependymoma is the fourth ventricle or adjacent ventricle wall. Less than one-third are found in the supra-tentorial region and these are more common in adults. Of those supra-tentorial ependymomas, extra-axial ependymomas are very rare [1]. We want to introduce our supra-tentorial and extra axial ependymoma case because it's very rare and mimicking a convexity mengioma which is very common.
CASE REPORT
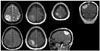
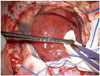
A 33-year-old woman without any medical history presented with 2 months of tingling and paresthesia on left extremity. After thorough neurologic examination, she suffered mild subjective left side weakness and falling tendency. Computed tomography (CT) demonstrated a hypo-dense tumor adjacent to dura at right parietal convexity (not shown). The mass was heterogenous but a contrast-enhanced magnetic resonance imaging (MRI) showed 41×42×29 mm sized extra-axial mass on right parietal convexity (Fig. 1). Which seems to be originated from meninges, but dural-tail sign wasn't significant and mainly slight hyperintense on T2-weighted image and hypointense on T1 with heterogenous enhancement. Under radiologic diagnosis of meningioma, we performed craniotomy and tumor removal. Under navigation-system guidance, dura was excised 1 centimeter from tumor margin. But, unlike meningioma, mass was completely separated from dura mater, and bone above the dura showed no evidence of invasion (Fig. 2). The mass was vascular, reddish, rubbery in consistency, and extra-axial with focal adhesion to arachnoid mater. After arachnoid dissection, tumor was totally removed, and arachnoid was coagulated where the mass was attached.
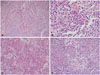
Histopathologically, the tumor was composed of monomorphic cells with high cell density and delicate capillary blood vessels. Tumor cell showed clear perinuclear halo due to cytoplasmic clearing and bland cytology. Mitotic count was 7 per 10 high power fields in the mostly active area. Multifocal necrotic areas, microvascular proliferation and hyalinizing areas were noted in the part of tumor. The perivascular pseudorosettes were vaguely found in the focal area (Fig. 3).
Immunoreactivity of GFAP was weakly positive and S-100 showed multifocal positivity. EMA immunostaining showed also focal dot-like positivity. OLIG2 expression was sparse (Fig. 4). Ki-67 proliferation index was 7.21% (not shown). Direct sequencing for isocitrate dehydrogenase 1 (IDH1) and IDH2 was the wild type. There was no 1p19q codeletion in fluorescence in situ hybridization (FISH) analysis. These findings were consistent with extra-axial clear cell ependymoma, grade III by WHO.
Post-operatively, the patient fully recovered and discharged. A post-operative CT and MRI revealed no sign of remnant tumor or recurrence. Due to complete resection of the tumor, the patient is followed monthly to check if it recurs, so we didn't perform radiotherapy.
DISCUSSION
Ependymoma is a tumor that arises from the ependymal lining of the ventricular system and central canal of the spinal cord. Usually, in pediatric cases the location of the tumor is intracranial, while in adults it is spinal. When occurred intracranially, they are more common in the infratentorial region, but some occurs supratentorially especially in adults [2]. Those which occur intracranially are known as cortical ependymomas and occurs in the parenchyma and lack of any evidence to support relation with the ventricular system. Extraaxial ependymoma, like our patient, is very rare. And also those who have clear-cell components are more rare than other subtypes which are identified as WHO grade III ependymomas [3]. To now, there is no definite agreement about the origin and management of these lesions. Also mechanism for the development of these neoplasms has not been identified. One of current theory is that subcortical or sub-ependymal mass extends extra-axially while growing [4]. Necrosis and calcification then follow and become extra-axial ependymoma. Another theory is the heterotopic placement of ependymal cell rests in the falx and the parasagittal region during fetal development with subsequent tumor growth. Ependymal rests are small bands of ependymal cells that extend out through the brain parenchyma from regions of sharp ventricular angulation [1].
There are significant clinical and radiographic differences between supratentorial and infratentorial ependymoma. Those who have infratentorial ependymoma commonly present with hydrocephalus whereas supratentorial ependymoma cause headaches, seizures, and focal neurological deficit [5]. Cortical ependymomas are usually small in size, and present with seizures alone unlike others [6].
Radiographically, supratentorial ependymomas tend to have cystic components and other features like calcification, necrosis and hemorrhage are common to both. Astrocytoma, supratentorial primitive neuroectodermal tumor, oligodendroglioma, and ganglioglioma can mimick similar feature radiographically [2]. But in our patient, the lesion mimicked a convexity meningioma.
Surgical total resection is the treatment of choice for supratentorial ependymoma [6]. And the role of radiotherapy in WHO grade III ependymomas has been established in earlier studies [4]. Roncaroli et al. [6] concluded that cortical lowgrade supratentorial ependymomas should be treated with surgery alone. Prognosis can be determined by the grade of the tumor and extent of surgical resection [7].
In our patient, even though she was diagnosed with clear-cell type ependymoma with the presence of necrosis and calcification which estimates WHO grade III ependymoma, pathological findings revealed that the tumor had relatively low proliferation profile (Ki-67 index 7%), so grading of the tumor with WHO grade III wasn't confident. Commonly, immunoreactivity of GFAP and S-100 are usually observed in ependymomas but, in the present case, immunoreactivity of GFAP was weak but positive, S-100 was also positive and OLIG-2 was negative. So pathologic findings of our case were not typical but consistent with enpendymomas.
Considering patient's age, tumor being gross totally removed, and pathological findings, we planned to observe her with caution and not perform radiotherapy. As observed by Mansur et al. [8], surgical removal extent of the tumor was the most important factor for the outcome. Mansur et al. [8] also observed that half of ependymoma patients experienced recurrence, so radiotherapy should be considered, despite the lack of data. But in spite of this data, regarding patient's young age and good performance state lead us to observe her carefully and perform radiotherapy when recurred because of possible side effects of radiotherapy. Prophylactic whole-spinal axis irradiation offers little utility in patients with localized supratentorial ependymomas, regardless of the tumor grade, because of the low incidence of these tumors seeding the spinal space [7].
Ependymomas can occur other than the central nervous system (CNS) like sacrococcygeal area, filum terminale, cauda equine, skin, subcutaneous tissue, presacral, pelvic, or abdomen [9]. Surgical total excision if possible is the best treatment option for extra-CNS ependymomas. Adjuvant radiotherapy might be needed when incomplete resection or recurrence. Chemotherapy has no evidence of effectiveness at all [10].
For prognosis, various factors are considered for ependymomas, including age, tumor location, histology, and extent of resection [1]. Most studies show that there is a survival advantage with gross total resection [1].
Extra-axial ependymoma is rare, and hard to be diagnosed radiographically due to low incidence. Even if total resection has been accomplished, close long-term follow-up must be needed because of high probability of recurrence.

 XML Download
XML Download