

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Central neurocytoma (CN) was first described in the 1980’s by Hassoun et al. [1] who studied two patients with intraventricular tumors using electron microscopy. CN is a benign tumor of the central nervous system that is classified as a grade II tumor by the World Health Organization (WHO) [23]. A combination of treatments, such as surgery with adjuvant ra-diation, can be considered for CN despite its good prognosis [234]. Because of the tumor’s rarity and its elusive nature, only a limited number of studies, case reports, and reviews have been published on CN.
EPIDEMIOLOGY
CN remains relatively rare, comprising about 0.1–0.5% of all brain tumors [5678]. CNs are most prevalent among young adults, and nearly 25% of all cases involve individuals in their thirties [49]. The age of affected individuals ranges from 8 days old to 67 years old with an overall median age of 34 years [610]. There is no correlation between gender and the incidence of CN [791112]. Some studies have indicated higher incidences of CNs in Korea, India, and Japan, which is possibly attributed to genetic differences among racial groups that make certain individuals more prone to CNs than others [81013141516171819202122232425262728]. The higher incidence in these Asian countries, make this tumor an important consideration when dealing with intraventricular tumors in these populations.
TUMOR LOCATION
CNs are neurocytomas located within the ventricles. Most CNs are found in the anterior half of the lateral ventricle, although some have reported to be found in the third and fourth ventricles [293031]. The tumor is also usually attached to the septum pellucidum near the foramen of Monroe [3233]. The cellular origination of CN is unclear; however, various authors have suggested CN may develop from neuronal cells, neuronal progenitor cells, neuronal stem cells, and multipotent precursor cells [112129343536].
Neurocytomas can also occur extraventricularly, along the spinal cord or the brain parenchyma [10]. Extraventricular neurocytomas (EVN) have been found in the cerebral hemi-spheres, spinal cord, brainstem, thalamus, pons, amygdala, pineal gland, retina, and cerebellum [5103738394041424344454647484950]. EVN is also classified as a grade II tumor [3]. However, EVN has a wider morphological spectrum and a higher degree of either focal or diffuse ganglionic differentiation than CN [61038515253].
CLINICAL MANIFESTATIONS
CN may increase the intracranial pressure by obstructing the interventricular foramen, which can lead to hydrocephalus [3254]. Patients may also experience nausea, vomiting, headache, seizures, decreased consciousness, weakness, and memory or vision problems [47303854555657]. In rare cases, intraventricular hemorrhage may also occur [58]. Patients with EVN present with similar symptoms, in addition to weakness and numbness in the limbs [20425960]. These symptoms are typically present for approximately 3–6 months, although the duration of symptoms can vary from a few days to many years [29305561]. The duration seems to be mostly related to tumor location, and does not seem to be correlated to the aggressiveness of the tumor [4].
RADIOLOGICAL FEATURES
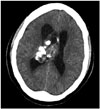
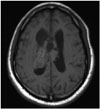
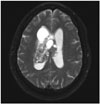
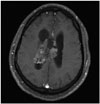
CN can appear as a dense mass in computerized tomography (CT) scans indicating calcifications, which occur in up to 50% of all cases, and present a patchy and coarse appearances (Fig. 1) [73033]. The tumor can also be heterogeneous because of the hypodense areas related to cystic degeneration [431]. In contrast-enhanced CT scans, CNs have mild to moderate enhancements [3262]. Furthermore, CNs appear slightly hypo-intense to iso-intense on T1-weighted magnetic reso-nance imaging (MRI) (Fig. 2), and iso-intense to hyper-intense on T2-weighted MRI (Fig. 3) [429313258]. In both T1- and T2-weighted MRI, hypointensity can indicate the presence of a hemorrhage, cyst, or calcification [31]. Typically, after a contrast agent is injected, moderate enhancement is seen on MRI for CN (Fig. 4) [295863]. Unfortunately, there is no estab-lished criterion to distinguish between CN and other tumors such as oligodendrogliomas on CT scans and MRI [41164].
SURGERY
Surgical management with a gross-total resection (GTR) is currently the gold standard treatment for CNs, which often has excellent prognosis and minimizes the chances of CN recurrence [65]. GTR is achieved in nearly 30–50% of all CN patients. In an analysis of 310 patients with CN who underwent a GTR, there was a 99% five-year survival rate [14546566]. In comparison, individuals who had surgery with only subtotal resection (STR) had an 86% five-year survival rate. STR of CN increases the rate of recurrence and decreases the rate of survival [65]. A recent multi-center study found that in 71 patients with CN, those with STR had a 3.8-fold higher risk of recurrence and adverse outcomes compared to patients with GTR [12]. For patients with STR, adjuvant radiotherapy was administered in this study.
RADIOTHERAPY
Fractionated radiotherapy
Radiotherapy and radiosurgery are non-invasive adjuvant treatments, but the toxicities from radiation are still being weighed against the benefits of tumor control [6567]. Because CNs usually have excellent prognosis when GTR is achieved, radiation is not always indicated [255455]. Radiotherapy and radiosurgery have been adopted as an adjuvant treatment when GTR cannot be achieved, the patient is inoperable, or the tumor is aggressive [1261].
A recent report suggests that fractionated radiotherapy (FRT) after STR had a statistically significant higher tumor control rate and improved survival in adults [1168]. A higher 5-year progression free survival has also been shown for patients who received adjuvant FRT after STR (67%) than patients without FRT (53%) [69].
Stereotactic radiosurgery
While FRT delivers multiple fractions of radiation in lower dosage, stereotactic radiosurgery (SRS) administers one higher dose (9 to 25 Gy) of radiation in 1-5 fractions. The first literature on the use of SRS for CN was published by Schild et al. [54]. SRS has been suggested to be potentially favorable to FRT. Although not statistically significant, Patel et al. [11] also reported that adjuvant SRS for patients with STRs demonstrated a 100% tumor control rate compared to an 87% tumor control rate for patients with adjuvant FRT [54].
Garcia et al. [70] also reported a higher tumor control rate of 93% with SRS versus 88% with FRT. The relative risk (RR) of SRS to FRT for recurrence was 0.57 less (95% CI: 0.21–1.57; log-rank p=0.85), and the RR for mortality was 0.23 less (95% CI: 0.05–1.05; log-rank p=0.22), although statistically insignificant. Lower complication was noted for patients with SRS, although distant tumor recurrence was slightly higher in patients who received SRS than those who received FRT. SRS is suggested to be at least as effective as FRT in achieving tumor control.
Prognosis
Schild et al. [54] reported a 5-year survival rate for patients who received FRT or SRS after surgical resection of 88%, while the 5-year survival rate for patients without adjuvant radiation was only 71%. Imber et al. [69] also found significantly improved survival rates when adjuvant radiotherapy is administered following STR. Patients with STR and FRT had a 67% 5-year survival rate, while patients with STR only had a 53% survival rate. Additionally, Kim et al. [71] reported that patients with STR had a lower recurrence rate after adjuvant therapy of FRT or SRS (1 of 12 patients) when compared to patients without adjuvant radiation (3 of 12 patients), although the difference was statistically insignificant. Overall, adjuvant therapy following incomplete resection of CNs appears to result in better tumor control.
Toxicity
Complications can arise from radiation therapy. A single institutional study found that 4 out of 7 patients who received FRT exhibited complications, such as white matter degradation or radiation necrosis, although FRT was effective in achieving local tumor control [71]. Chen et al. [72] found that among 60 patients treated with radiation therapy, 28 patients exhibited grade I neurotoxicity which resulted in short-term memory impairment and motor deficit. Seven patients displayed grade II neurotoxicity, while three patients had grade III neurotoxicity [72]. The associated symptoms included cognitive disturbance, hemianopsia, seizure, and involuntary movement [72].
CHEMOTHERAPY
Although chemotherapy is not a primary treatment modality for CN, chemotherapy has been used as an adjuvant or salvage therapy for recurrent CNs or inoperable patients [113173]. There are no studies using chemotherapy as a primary form of treatment for CN, nor a comparison of the efficacy between radiotherapy and chemotherapy as an adjuvant treatment [411737475]. Only a few case reports noted partial tumor regression following chemotherapy, and only one study reported a child with a complete response using a combination of topotecan, carboplatin, and phosphamide in three cycles [475767778].
HISTOPATHOLOGICAL ANALYSIS AND MOLECULAR PATHOGENESIS
CN has been relatively difficult to diagnose because of its histopathological similarity to other brain tumors, such as oligodendrogliomas and ependymomas [41164]. Light microscopy is ineffective in identifying CNs [97980]. Generally, immunohistochemistry is performed for the diagnosis of CN [92979].
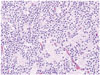
The histology of CN can vary throughout a single specimen and is typically benign (Fig. 5) [10]. The tumor cells create a "honeycomb pattern," and appear small and round with scant cytoplasm and stippled chromatin [410243132]. Since these characteristics are similar to the appearance of oligodendrogliomas, there is a potential opportunity for misdiagnosis [41011243132]. Likewise, the pathological features of ependymo-mas are similar to the perivascular rosette or straight line cell arrangements that are also seen in CN [10]. Therefore, multiple immunohistochemical markers are helpful in differentiating CNs from other tumors.
Although less commonly used, electron microscopy can be another helpful tool in diagnosing patients with CNs by looking for parallel arrays of microtubules with dense-core neurosecretory granules and clear vesicles [7212436].
Immunohistochemical markers
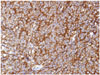
Synaptophysin is one of the major molecular markers for CN [10]. Positive staining for synaptophysin, a transmembrane glycoprotein present in presynaptic vesicles of neurons, is a strong indicator for neuronal cells and its neoplasms (Fig. 6). Synaptophysin staining is usually found in the fibrillary and perivascular areas of CN [98182].
In addition to positivity for synaptophysin, negativity for neuron specific enolase (NSE) and vimentin has been reported to suggest CN over oligodendroglioma and clear cell ependymoma (Table 1) [4]. NSE is a glycolytic enzyme located in the cytoplasm of neurons [83]. Although it is present in CN, it lacks neuronal specificity and has been reported to be present in non-neuronal neoplasms [924]. Vimentin is an intermediate filament protein found in glial cells, and is usually present in oligodendrogliomas and clear cell ependymomas, but absent in CN [8485].
Epithelial membrane antigen (EMA) is another protein that differentiates CN from oligodendroglioma and ependymoma. EMA, which is normally expressed in epithelial cells, is present in ependymal cells in the central nervous system, as well as in ependymomas [86]. Furthermore, EMA positivity has also been linked with other glial tumors such as glioblastoma, astrocytoma, and oligodendroglioma [8687]. Thus positivity of EMA can suggest ependymoma and oligodendroglioma over CN [4].
Neuronal nuclei (NeuN) is present in the nuclei and perinuclear cytoplasm of post-mitotic neurons in the central nervous system. Positive staining for NeuN suggests the neuronal nature of neoplasms and is considered to be a reliable marker for clear cell neoplasms of the central nervous system, which include CN, oligodendroglioma, and clear cell ependymoma [48889]. It has also been reported that positive staining for NeuN correlates with a lower proliferation index [490].
Glial fibrilllary acidic protein (GFAP), which is detected in glial cell tumors, is usually absent in CN. It is the most abundant intermediate filament protein in astrocytes, and is usually present in astrocytes that infiltrate or surround CN [4215152539192]. Cases of CN with GFAP positivity suggest glial differentiation of bipotential (astrocytic and neuronal) precursor cells, and also correlates with a more malignant disease course [93].
Neurofilament (NF), which exists as intermediate filaments in neurons, is largely absent in CN. This suggests that the full differentiation of CN cells to developed neurons is rare [1234]. Vasiljevic et al. [12] reported that positivity in NF is a key diagnostic difference between CN and pineal parenchymal tumor.
Oligodendrocyte transcription factor 2 (Olig2), which is a transcription factor that regulates oligodendroglial differentiation, is also generally absent in CN. Olig2 can be used as a diagnostic marker for oligodendroglioma, and positivity for Olig2 suggests oligodendroglioma over CN [9495]. This also argues the rarity of CN cells undergoing glial differentiation.
Chromagranin A (chrA) is a neuroendocrine protein located on secretory vesicles of neurons. ChrA is generally absent in CN, but cases of positivity have been reported [969798]. Peng et al. [96] suggested that the positivity of chromogranin A may be due to the presence of ganglion cells in CN. Overall, ChrA is not a reliable marker for the diagnosis of CNs; however, ChrA positivity in some CNs may provide an insight to the cellular and developmental origins of CN [98].
Pathology (Ki-67)
The MIB-1 Labeling Index (MIB-1 LI) is an important prognostic tool for CN (Fig. 7). In 2015, Imber et al. [69] measured progression-free survival (PFS) by Kaplan-Meier and Cox proportional hazards methods, and found that the two year PFS was 48% for MIB-1 LI >4%, and 90% for MIB-1 LI <4%. Similarly, CN with MIB-1 LI <2% had a 10-year survival rate of 90%, compared to MIB-1 LI >2%, which had a 10-year survival rate of 63%.
MIB-1 LI has also been reported to be an indicator of tumor relapse. It has been reported that CN with MIB-1 LI >2% had a 63% chance of recurrence; whereas CN with MIB LI <2% had only a 22% chance of recurrence over a 150-month period [499]. Furthermore, Chen et al. [93] found that out of the nine patients presented in the study, the four that experienced tumor recurrence or death from continuous tumor growth or surgical complications had MIB-1 LI >2%, suggesting that an MIB-1 LI >2% may indicate a more aggressive disease course.
MIB-1 LI may also be used to determine the tumor grade. Sharma et al. [18] reported that the only proliferation marker that correlated with CN atypia is the MIB-1 LI. Söylemezoglu et al. [52] found that a MIB-1 LI >2% correlated with microvascular proliferation and suggested that a CN with MIB-1 LI >2% should be termed ‘atypical’ [4]. Atypical CN are also known to spread through the cerebrospinal fluid and metastasize in the ventricles or the spinal cord [100101102103].
Genetic alterations
Many types of genetic mutations have been associated with CNs. N-myc proto-oncogene (N-Myc), which is an oncogene associated with the development of other cancers such as neuroblastoma and medulloblastoma, is overexpressed in CN [34104105106]. The overexpression of N-Myc in neuroblastoma seems to indicate a poorer prognosis [34107]. N-Myc is required for neural proliferation, but inhibits complete neural differentiation of neuronal progenitor cells [34108]. The levels of N-Myc are inversely related to the tumor suppressor gene encoding Myc box-dependent-interacting (BIN-1) protein, which has been found to be significantly underexpressed in neurocytomas, as well as other tumors [34105]. This suggests that CN may contain a mutation somewhere in the pathway that includes both N-Myc and BIN-1, and that the respective overexpression and underexpression of N-Myc and BIN-1 may play a large role in CN tumorigenesis [34105].
Phosphatase and tensin homolog (PTEN) gene is a tumor suppressor gene also found to be overexpressed in CN [105]. Musatov et al. [109] showed that PTEN overexpression inhibits neural differentiation in PC12 cells by phosphoinositide 3-kinase and mitogen-activated protein kinase pathways [34]. Thus, PTEN and N-Myc overexpression together could po-tentially warrant the lack of full neuronal differentiation frequently seen in CN.
In addition, Sim et al. [35] found that insulin-like growth factor 2 was overexpressed in CN cells compared to cells in the ventricular zone, and may play a key role in the proliferation of neurocytoma, similar to its role in the proliferation of glioblastoma multiforme [34110].
Similarly, platelet-derived growth factor D (PDGF-D) and neureglin 2 (NRG-2) were found to be overexpressed in CN. PDGF-D overexpression has been found to be involved with the maturation of certain tumors [34111], whereas NRG-2 overexpression has been linked to the proliferation of neuroblasts, as well as the aggressiveness in breast carcinoma [34112113]. Overall, PDGF-D and NRG-2 can also offer an explanation for the tumorigenesis of neuronal progenitor cells [34].
CONCLUSION
CN is a benign tumor of the CNS that has an excellent prognosis. Surgery with gross total resection is the most preferable, correlated with the best long-term survival rates and local tumor control. Adjuvant radiotherapy may be considered for residual CN following STR, large CN size, or CNs near inoperable regions. Radiotherapy or chemotherapy the primary treatment for CNs has not been thoroughly examined.
Many histological markers are available for the diagnosis of CN, although some markers are also sensitive to other tumors. The MIB-1 LI is currently the most accurate tool to determine prognosis, tumor relapse, and tumor grade. Further molecular and genetic studies may offer insights into other immunohistochemical methods for improved diagnostic accuracy.


 XML Download
XML Download