

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Subependymal giant cell astrocytoma (SEGA) is a benign slowly growing tumor, which typically arises at the caudothalamic groove adjacent to the foramen of Monro and is composed of large ganglioid astrocytes [1]. Tumors are pathologically classified as grade I glioma by the World Health Organization (WHO). Several authors suggest the term "subependymal giant cell tumor", but most authors still use the term SEGA. These tumors are diagnosed with symptoms of elevated intracranial pressure, obstructive hydrocephalus, and focal neurological deficits [2]. The SEGA is the one of the major features in the diagnostic criteria for the tuberous sclerosis complex (TSC) [3]. Even if there is a debate whether SEGA occurs outside the TSC, 10% to 20% of the TSC patients develop SEGA, resulting in the major cause of the TSC-related morbidity and mortality during the pediatric age [1,4].
An operation is indicated in any SEGA that is increasing in size or causing symptoms. We reviewed the clinical characteristics of five cases of the pathologically proven SEGA in our hospital.
CASE REPORT
Clinical analysis of five patients
Between May 1997 and July 2012, five patients of SEGA were histologically diagnosed with grade I according to the WHO classification. The pathology showed the pleomorphic multinucleated eosinophilic tumor cells with abundant cytoplasm, and these elongated tumor cells formed streams (Fig. 1A). The tumor cells were clustered and arranged in perivascular pseudopalisading pattern (Fig. 1B). The Ki-67 labeling index was <1%, and the tumor cells were immune-positive for the glial fibrillary acidic protein and microtubule-associated protein 2 (Fig. 1C). The radiological follow-up was performed for at least two years. Four patients were men and one was female. The median age was 18 years (range, 8 to 26). The clinical symptoms were presented as seizure in two patients and headache in three patients. Radiologically, all five patients presented with the mass of lateral ventricle near the foramen of Monro. The median size of tumors was 2.5 cm (range, 1.9-4.0). Two patients showed the solitary lesion. One patient had the associated subependymal nodules, and two patients were associated with subependymal nodules and cortical tubers. Two patients were associated with hydrocephalus. The clinical information is listed in Table 1.
The median follow-up duration was 7.4 years (range, 2.0-14.3). The TSC was diagnosed with the updated diagnostic criteria for TSC 2012 [3]. Three patients were associated with the TSC, and one patient out of the three had a family history. Four patients showed the SEGA at their first presentation, and one patient experienced a 1.9 cm-sized growing mass during 7.7 years follow-up after the diagnosis of the TSC. The transcallosal approach and tumor removal were performed for all the patients. The mass was totally removed in four patients and subtotally in one. Postoperatively, one patient took the medication for seizure, which was controllable. Hydrocephalus was controlled without the shunt operation. The subtotally removed mass showed the postoperative recurrence 4.1 years later, and the recurred mass was stable for 4.5 years after the recurrence.
Case illustration
Case 2
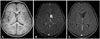
An 18-year-old man had headache for one month. The magnetic resonance images (MRI) and computed tomography (CT) of the brain displayed 3 cm-sized solitary lesion associated with cyst and calcification (Fig. 2A, B). The T1-weighted MR images showed an iso-signal intensity and T2-weighted images with high signal intensities. The brain CT showed enhanced mass after the contrast administration. The mass was subtotally removed by the transcallosal approach, because of the adhesion of hypothalamus (Fig. 2C). After the operation, he complained mild memory impairment, and the hydrocephalus was controllable without shunt operation. After 4.1 years, the radiological follow-up showed the 0.8 cm nodular enhancing lesion in the right side of the septum pellucidum (Fig. 2D). This lesion was stable for 4.5 years after the recurrence without any symptoms.
Case 4
An 8-year-old boy had the headache and seizure. He had a familial history of TSC 7.7 years ago, his brain MRI showed the 1 cm-sized mass adjacent to the foramen of Monro (Fig. 3A). There were multiple subependymal nodules on both lateral ventricle and multiple cortical tubers on the cerebral hemisphere. He took the medicine for the seizure. On admission, the brain MRI displayed 1.9 cm-sized enlarged lesion (Fig. 3B). The T1-weighted MR images showed an iso-signal intensity and T2-weighted images with a high signal intensity with the contrast enhancement. The mass was totally removed via the transcallosal approach without any neurological deficit. The seizure was controllable with the medication. There was no recurred lesion, and all multiple subependymal nodules and cortical tubers were stable for two years after the operation (Fig. 3C).
DISCUSSION
SEGA typically occurs in the lateral ventricle near the foramen of Monro and is often associated with features of the TSC. One study suggested that SEGA could be unique to the TSC [5]. The reexamination revealed no further examples of the SEGA in the patients without features of the TSC. TSC is a rare autosomal dominant genetic disease, and the nonmalignant tumors grow in multisystem such as brain, kidneys, heart, eyes, lungs, and skin [2]. The increased mammalian target of rapamycin (mTOR) activation leads to the disorganized cellular overgrowth, abnormal differentiation, increased protein translation, and formation of tumors. There are characteristic brain lesions such as cortical tuber, subependymal nodule, and SEGA. The brain lesions are occupied in 5% to 20% of the TSC [4]. The cortical tuber shows the high rate of epilepsy without oncologic growth potential. SEGA could be the transformation of subependymal nodules; however, this is controversial [2,6]. Even if the subependymal noduls and SEGAs have similar histopathological features, SEGAs are typically located in the caudothalamic groove as opposed to the subependymal nodules located in the ependymal lining of the lateral ventricles along the caudate nucleus [6]. The subependymal nodules showed calcified and nonenhancing lesions, whereas the SEGAs showed the contrast enhancement. However, the SEGAs grow, whereas the subependymal nodules remain stable in size. SEGAs are potentially lethal and have been shown to be responsible for 25% of the excess mortality caused by the TSC [7]. For the diagnostic purposes of the TSC, SEGAs are radiologically defined as a lesion at the caudothalamic groove with either a size of more than 1 cm in any direction or a subependymal lesion at any location that has shown serial growth on the consecutive imaging regardless of the size [3]. A growing subependymal lesion even in the absence of enhancement should be considered as a SEGA [3]. New SEGAs very rarely arise after 20-25 years of age [4]. Brain imaging should be recommended every 1 to 3 years until the age of 25 years [1]. Because of a lack of knowledge of the SEGA growth behavior beyond 25 years of age, the interval of radiological follow-up may be prolonged in the presence of a stable lesion. In this study, three patients were associated with the TSC, and one patient did not associate with TSC even after the reexamination. One patient did not have enough information to diagnose the TSC. All the patients showed indolent clinical behavior even after the recurrence.
Despite the pathological presence of the focal necrosis and mitoses of the SEGA, clinical follow-up studies revealed a lack of aggressive tumor behavior after the surgery alone [8]. There is a discrepancy between the histological and clinical features, and the presence of mitosis and necrosis does not lead to unnecessary radiotherapy or chemotherapy after the surgery. However, even if SEGA are clinically benign tumors, there was a rare case report with the spinal metastases and increased Ki-67 labeling index [9]. The intracranial hemorrhage of SEGA had been rarely reported [10].
The treatment of SEGAs has been solely surgical [1]. Although benign and typically slow growing, they can cause serous neurological compromise including the obstructive hydrocephalus. The exact surgical time is still controversial, but the treatment is indicated for a new set of symptoms or radiological evidence of the tumor growth [1,3]. Small tumors are usually less invasive and associated with excellent clinical outcomes with low morbidity and mortality [11]. However, at a later stage, the tumor invades neighboring structures such as the fornix, hypothalamus, basal ganglia, and genu of internal capsule, which are associated with a higher surgical morbidity and mortality. The SEGAs can be removed as soon as clear evidence of the growth is confirmed [11]. A postoperative radiotherapy is not recommended, because it may be associated with an increased risk of the secondary malignancy [12]. In this study, the median size of tumors was 2.5 cm (range, 1.9-4.0), and the masses were removed without the neurological deficit. We also suggested the surgical removal in the cases of growing mass to avoid severe neurological deficits.
The total surgical resection cannot be possible for the bilaterally located SEGAs or invasive lesions to the neighboring structures. The medical treatment is favored in the case of multiple tumors and lesions for which gross total resection in unlikely and growing residual tumors [1]. The recent clinical studies, the mTOR inhibitor everolimus, significantly decrease the volume over 50% of SEGAs in 35% to 42% at six months of the treatment [13,14]. Even if the side effect is insignificant, mTOR inhibitor could be associated with the stomatitis and upper respiratory tract infections. In addition, the cessation of the treatment may result in tumor regrowth [15].



 XML Download
XML Download