

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Immunoglobulin G4 (IgG4)-related disease is a newly-recognized fibrosing inflammatory disease characterized by a dense lymphoplasmacytic infiltrate rich in IgG4 positive plasma cells, storiform fibrosis, obliterative phlebitis, and, often but not always, elevated serum IgG4 concentrations [1]. The disease was first defined in relation to the pancreas but many other sites of involvement have been progressively recognized based on similar histopathological features [2]. In the central nervous system, IgG4-related disease was initially demonstrated in the form of hypophysitis and has been recently described in a subset of cases previously diagnosed with idiopathic hypertrophic pachymeningitis [3,4,5]. However, the IgG4-related disease involving bone is extremely rare. Here, we report a case of IgG4-related hypertrophic pachymeningitis with skull involvement.
CASE REPORT
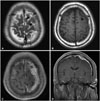
A 65-year-old man with a history of hypertension presented to the emergency room following a generalized tonic seizure that lasted for 10 minutes. His consciousness was restored shortly before arrival and his neurological examination was unremarkable. Brain computed tomography revealed a slightly hyperdense mass at the left frontal convexity, without adjacent bone abnormalities. Brain magnetic resonance (MR) imaging demonstrated an irregular and sheet-like 2.4 cm diameter extra-axial mass at the vertex of the left frontal area. T2- and T1-weighted MR images showed a hypointense mass, and contrast enhanced T1-weighted MR imaging revealed a homogenously enhanced mass with diffusely thickened and enhanced dura mater along the left hemisphere. In addition, the skull adjacent to the mass was enhanced, and there was no skull defect or hyperostosis (Fig. 1). We initially suspected hypertrophic cranial pachymeningitis (HCPM) and investigated its known causes.
Routine laboratory evaluation revealed normal blood counts, erythrocyte sedimentation rate, and chemistry. Lumbar cerebrospinal fluid (CSF) study showed clear fluid and normal values for cell counts, protein, and glucose. Examination of the CSF for mycobacteria, fungi, and malignant cells was negative. A work up for sarcoidosis, collagen vascular disease, and systemic malignancy was also negative. Consequently, the left frontal bone with enhancement and enhanced mass were excised. On the operative findings, although it was uncertain whether the mass invaded the involved overlying skull, the mass originated from the dura and grew in. The consistency of the mass was solid, rubbery, and gray in color.
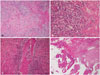
Histopathological findings of the mass demonstrated diffuse inflammatory cell infiltration, especially numerous plasma cells and lymphocytes in the sclerotic background (Fig. 2A, B). And involved skull also revealed lymphoplasmacytic infiltration and fibrosis (Fig. 2D). We consequently diagnosed idiopathic hypertrophic intracranial pachymeningitis and started steroid treatment. Four months after surgery, follow-up MR imaging revealed thinning and shrinking of the enhanced dura.
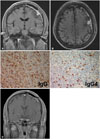
Eight months after surgery, a partial seizure of the right upper arm developed. Brain MRI revealed a new mass adjacent to the previous one, and diffuse dura enhancement. Recurrence was suspected, and re-excision was considered (Fig. 3A, B). However, we decided to perform additional immunohistochemical staining of immunoglobulin G (IgG) and IgG4 for the previous specimen, because it was suggested that an IgG4-related sclerosing disease might represent a component of idiopathic hypertrophic pachymeningitis. Immunohistochemical studies revealed increased both IgG4 (+) plasma cells (66/HPF) and IgG4+/IgG+ cell ratio (57% in the hot spot) (Fig. 3C). Also obliterative phlebitis was further recognized (Fig. 2C). The histological features of dense lymphoplasmacytic infiltration, fibrosis and obliterative phlebitis in conjunction with the immunohistochemical results support the diagnosis of highly suggestive of IgG4-related hypertrophic pachymeningitis. But patient's serum IgG4 concentration is not elevated. Then the patient took continuous oral prednisolone and immunosuppressants such as either azathioprine or cyclophosphamide. Brain MRI obtained at 3 months after immunosuppressant medication demonstrated that the enhanced mass and thickened dura had disappeared (Fig. 3D). The patient has since been doing very well, without any significant symptoms suggestive of disease recurrence.
DISCUSSION
Hypertrophic cranial pachymeningitis is characterized by marked diffuse thickening of the cranial dura mater. Numerous clinico-pathological entities cause thickening of the pachymeninges. Despite evaluation for the cause of the HCPM, there are some cases without cause, termed idiopathic hypertrophic cranial pachymeningitis (IHCPM). IHCPM is a diagnosis of exclusion and a dural biopsy is essential to exclude known causes such as infectious, neoplastic, or autoimmune disorders. Histopathological findings compose of a thick fibrous dura often associated with chronic inflammatory cell infiltration consisting of lymphocytes and plasma cells. Granulomatous findings have been noted in about 10% of the reported cases of HCPM [6].
Recently, however, Chan et al. [3] proposed that a proportion of IHCPMs might be a part of the IgG4-related disease spectrum. IgG4 was first recognized as being associated with sclerosing disease in 2001 when Hamano et al. [7] reported that patients with autoimmune pancreatitis had elevated serum levels of IgG4 in comparison to patients with other causes of chronic pancreatitis. Since then, IgG4 has been recognized to be associated with diseases involving numerous organs. In the central nervous system, the IgG4-related disease has been initially demonstrated in the form of hypophysitis and some cases of IgG4-related hypertrophic pachymeningitis have been reported in the literature [3,4,5]. However, IgG4-related hypertrophic pachymeningitis involving the skull is extremely rare. To our knowledge, Lin and Lai [8] reported the first case of an IgG4-related hypertrophic pachymeningitis with skull hyperostosis. Our case is the second involving the skull, and the difference with the first case is that there was no hyperostosis in our case.
Radiologically and histologically IgG4-related hypertrophic pachymeningitis shares many similarities with IHCPM. On MRI, it is characterized by a marked thickening of the dura that enhances along its edges. The characteristic histological features are extensive lymphoplasmacytic infiltrate, whorling-pattern fibrosis and obliterative phlebitis [9]. However, IgG4-related hypertrophic pachymeningitis is confirmed by the immunohistochemical analysis of more than 10 IgG4 bearing plasma cells/HPF and, often but not always, elevated serum IgG4 concentrations [2]. And most importantly IgG4+/IgG+ plasma cell ratio >40% is required for diagnosis. The present case indicates that the HCPM can be misdiagnosed as IHCPM unless IgG4 immunostaining was performed.
Although no randomized treatment trials for IgG4-related disease have been conducted, glucocorticoids are typically the first line of therapy. In addition, azathioprine, mycophenolate mofetil, and methotrexate are frequently used as glucocorticoid-sparing agents or remission-maintenance drugs after glucocorticoid-induced remissions [1]. In our case, the patient was initially treated with steroids, and then immunosuppressant treatment with azathioprine and cyclophosphamide was initiated. Eight months after the start of immunosuppressant treatment, the patient showed no adverse events or complications.
It is difficult to know the natural history of IgG4-related hypertrophic pachymeningitis because the disease is rare and recently diagnosed. From the perspective that IHCPM is clinically similar with IgG4-related hypertrophic pachymeningitis, it may be possible to infer the natural history of IgG4-related hypertrophic pachymeningitis. Parney et al. [10] reported the outcomes of 33 patients with IHCPM such that 26% experienced full remission without steroid dependence, 15% experienced steroid-dependent partial or complete remission, 15% experienced a progressive course in spite of steroid therapy, and 32% died regardless of treatment. However, combined extracranial diseases are important to the overall clinical course of IgG4-related disease. To reveal the natural history of IgG4-related meningeal disease, the work-up of systemic diseases and long term follow-up should be performed.
In conclusion, IgG4-related hypertrophic pachymeningitis is very rare and only one case with skull involvement has been reported in the literature. Here, we report the second IgG4-related hypertrophic pachymeningitis case with skull involvement. It is difficult to confirm IgG4-related hypertrophic pachymeningitis, unless IgG4-special staining is performed after excision for HCPM. Therefore, IgG4-related hypertrophic pachymeningitis should be considered in the differential diagnosis of HCPM.
 XML Download
XML Download