

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Epigenetics can be defined as mitotically heritable changes in gene expression that are not due to changes in the primary DNA sequence. Epigenetic mechanisms, including those involving enzymatic modifications to DNA or histone proteins, thereby regulating gene expression, are increasingly recognized as a source of phenotypic variability in biology. The discovery of altered epigenetic profiles in human neoplasia has been a major factor in constructing a new paradigm, in which epigenetic variability contributes significantly to human disease. Because of their reversible nature and their role in gene expression and DNA structure, epigenetic alterations, especially those related to changes in histone acetylation, are a current focus for therapeutic drug targeting in clinical trials.
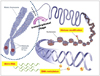
Covalent modifications of DNA and amino acids on histones are two major mechanisms of epigenetic gene regulation (Fig. 1). First, DNA methylation results from the addition of a methyl group to cytosine thereby creating 5-methylcytosine. In mammals, this almost always occurs at the 5'-CpG-3' dinucleotide, though occasionally methylation is also observed at CpNpGs [1]. DNA methylation is controlled by DNA methyltransferases (DNMT) that create (DNMT3A, DNMT3B) or maintain (DNMT1) patterns of methylation [2]. DNA methylation is required to silence genes on the inactive X chromosome [3] or for the allele-specific expression of some imprinted loci [4]. Methylation is also required to silence transposable elements, to maintain genomic stability [5] and is a critical regulator of genes that contribute to cell pluripotency [6].
Another major epigenetic mechanism is the post-translational modification of the N-terminal tails of histone proteins by acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, ADP ribosylation, biotinylation and other potential modifications [7]. Several families of enzymes catalyze post-translational modifications of histones, including acetyltransferases and deacetylases, methyltransferases and demethylases. Additionally, multiple types of modifications can take place on a single histone molecule, increasing combinatorial complexity. In addition, each amino acid residues can be modified in different states, such as mono-, di-, or tri-methylation at lysine residues.
In addition to DNA methylation and histone modifications, there are other potential epigenetic mechanisms that include specific deposition of histone variants, noncoding RNAs, chromatin remodeling, or nuclear organization of DNA. The interplay between histone modifications and other chromatin modifications leads to the dynamic regulation of chromatin structure and thereby affects several relevant cellular processes including transcription, DNA replication, DNA repair, and genomic stability [8]. Together, these add additional layers to the regulation of gene expression in both normal and diseased states.
HISTONE MODIFICATIONS
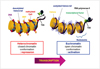
Chromatin is the condensed combination of DNA and histones within the nucleus of a cell. The structural and functional unit of chromatin is the nucleosome, which consists of a disc-shaped octamer composed of two copies of each histone protein (H2A, H2B, H3, and H4), around which 147 base-pairs of DNA are wrapped twice (Fig. 2A) [9]. Electron microscopy studies have revealed that organization of nucleosomal arrays structurally resembles a series of "beads on a string", with the "beads" being the individual nucleosomes and the "string" being the linker DNA [3]. Linker histones, such as histone H1, and other non-histone proteins interact with the nucleosomal arrays to further package the nucleosomes into higher-order chromatin structures [8].
Histones are highly conserved across species. These proteins contain a conserved globular domain, which mediates histone-histone interactions within the octamer (Fig. 2A) [9]. In addition, there are two small tails protruding from the globular domain: an amino (N)-terminal domain, constituted by 20-35 residues that are rich in basic amino acids, and a short, protease accessible carboxy (C)-terminal domain [9]. Histone H2A is unique among the histones due to its possession of an additional 37 amino acids in the carboxy-terminal domain that protrudes from the nucleosome [9]. Additional histone variants have been identified [10].
In particular, their tails can be subject to a remarkable number of modifications, although examples of modifications within the globular domain have also been identified. Histone modifications include acetylation, methylation and phosphorylation, but also some less-studied modifications such as ubiquitylation, sumoylation, ADP ribosylation, deamination and proline somerization (Fig. 2B) [9,11,12]. Each of these histone modifications directly or indirectly affect chromatin structure, thereby leading to alterations in DNA repair, replication and gene transcription (Table 1). The effect of histone modifications on gene transcription can broadly be categorized into active versus passive marks. Moreover, numerous studies have reported the presence of site-specific combinations and interdependence of different histone modifications, which may be interpreted as the so-called "histone code". The role of histone modifications and their crosstalk in different cellular processes will be described in the following two sections. In particular, I will focus on discussing the two well-studied histone marks: histone acetylation and methylation [12,13].
Histone acetylation
Acetylation is a reaction based on the introduction of an acetyl functional group into a chemical compound. More specifically, it is a reaction between the hydrogen atom of a hydroxyl group and an acetyl group (CH3CO). Lysine acetylation refers to the α-amino group of a lysine residue, which is a reversible modification referred to as Nε-acetylation. Nε-acetylation is different from the Nα-acetylation, which is an acetylation of the N-terminal α-amine of proteins and a common modification in eukaryotes [14].
In 1964, Allfrey first discovered that histone proteins exist in an acetylated form [14]. Lysine acetylation is controlled by two types of histone acetyltransferases (HATs), which require acetyl-CoA to be specifically recognized and bound by the Arg/Gln-X-X-Gly-X-Gly/Ala segment of HATs, which then allows for the transfer of an acetyl group to the α-amino groups on the N-terminal tails of histones [15]. Histone deacetylases (HDACs) reverse this modification [16]. Soon after the discovery of the first HATs and HDACs in the mid-1990s, identification of a large number of HATs followed, resulting in a surge of interest in histone acetylation. Most HATs come in the form of large, multi-protein complexes. The different components of HAT complexes ensure locus targeting and chromosomal-domain and substrate-specificity. Based on sequence similarity, HATs can be organized into families, which seem to exhibit different mechanisms of histone-substrate binding and catalysis [17]. The dynamic equilibrium of lysine acetylation in vivo is governed by the opposing actions of HATs and HDACs. Similar to acetyltransferases, the HDACs are also part of large, multi-protein complexes [18].
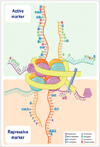
Lysine acetylation is believed to neutralize the positive charge of histone tails, weakening histone-DNA [19] or nucleosome-nucleosome interactions [20], and inducing an open (euchromatin-like) conformational change (Fig. 3) [21]. This results in destabilized nucleosomes and chromatin structure, thus facilitating access to the DNA for different nuclear factors, such as the transcription complex. Conforming to this model, hyper-acetylation of histones is now considered a hallmark of transcriptionally active chromatin. Deacetylation of histones by HDACs results in a decrease in the space between the nucleosome and the DNA, leading to a closed (heterochromatin-like) chromatin conformation that diminishes accessibility for transcription factors (Fig. 3).
Histone methylation
Protein methylation is a covalent post-translational modification that commonly occurs on carboxyl groups of glutamate, leucine, and isoprenylated cysteine, or on the side-chain nitrogen atoms of lysine, arginine, and histidine residues [22]. As described in 1964, histones have long been known to be substrates for methylation [23]. For histones, methylation occurs on the side chain nitrogen atoms of lysines and arginines. The most heavily methylated histone is histone H3, followed by histone H4.
Arginine can be either mono- or di-methylated, with the latter in symmetric or asymmetric configuration [24]. Protein arginine methyltransferases (PRMTs) are the enzymes that catalyze arginine methylation. PRMTs share a conserved catalytic core but are very different on the N- and C-terminal regions, which likely determine substrate specificity [25]. There are two types of PRMTs: type I enzymes catalyze mono and asymmetric di-methylation of arginine and type II enzymes catalyze mono- and symmetric di-methylation of arginine [26]. Several studies have suggested that certain arginine methyltransferases, such as PRMT5, may repress the expression of genes involved in tumor suppression [26].
Similar to arginine methylation, lysine methylation can occur in mono-, di-, and tri-methylated forms. Some of the lysine residues methylated in histones H3 and H4 are also found to be substrates for acetylation. The enzymes that catalyze methylation on lysine residues have been grouped into two classes: lysine-specific, SET [Su(var), Enhancer of Zeste, and Trithorax] domain-containing histone methyltransferases (HMTs) share a strong homology with a 140-amino acid catalytic domain known as the SET domain, and non-SET containing HMTs. It is important to note that not all SET domain-containing proteins are HMTs nor are all HMT activities mediated by SET domains [27].
The consequences of lysine methylation are extremely diverse. Depending upon a particular lysine, methylation may serve as a marker of transcriptionally active euchromatin or transcriptionally repressed heterochromatin [28]. For instance, methylation of histones H3K9, H3K27, and H4K20 are mainly involved in formation of heterochromatin (closed chromatin conformation). On the other hand, methylation of H3K4, H3K36, and H3K79 are correlated with euchromatin (open chromatin conformation) (Table 2). Moreover, it seems that H3 clipping, a mechanism involving the cleavage of 21 amino acids of histone tails following the induction of gene transcription and histone eviction, occurs on histone tails that carry repressive histone marks [29].
Until very recently, the dogma was that methylation was an irreversible process. With the identification of the first lysine demethylase, lysine-specific demethylase 1 (LSD1) in 2004, the view of histone methylation regulation became much more dynamic, opening the way for identification of many more histone demethylases [30]. LSD1 demethylates both mono- and di-methylated K4 on H3 [30]. In 2006, the protein JHDM1A was identified as the first jumonji-domain-containing histone demethylase that removes methyl groups from mono- and di-methyl H3K36 [31]. The jumonji (JmjC)-domain-containing proteins belong to the deoxygenase superfamily and use a demethylation mechanism distinct from that of LSD1/KDM1 [32]. These enzymes can demethylate trimethylated lysine residues. The JMJD2/KDM4 demethylases are tri-methyl demethylase families that were reported soon after the first JHDM1A was discovered [33]. Over the past few years, a series of studies have identified additional jumonji-domain-containing families that have methylated substrates K4, K9, K27, and K36. Despite the tremendous and exciting progress in the last few years, the field of histone demethylases is still in its early days and our knowledge of the biological role of these enzymes is still rather limited.
Histone code
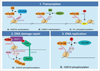
Over the past few years, the field of epigenetics has provided a great deal of evidence arguing that histone modifications act in a combinatorial and consistent manner, leading to the concept of the "histone code" (Table 3) [34]. Different histone modifications present on histone tails generate a "code", which can be read by different cellular machineries thereby dictating different cellular outcomes, such as activation or repression of transcription, DNA replication, and DNA repair (Fig. 4) [34]. The histone code hypothesizes that the transcription of genetic information encoded in DNA is in part regulated by chemical modifications to histone proteins, primarily on their unstructured ends. Along with similar modifications such as DNA methylation, it is part of the epigenetic code. Many of the histone tail modifications are associated with chromatin structure, and both histone modification state and chromatin structure are correlated with gene expression levels. The histone code hypothesis is that histone modifications serve to recruit other proteins by specific recognition of the modified histone via specialized protein domains, rather than through simply stabilizing or destabilizing the interaction between a histone and the underlying DNA. These recruited proteins then act to actively alter chromatin structure or to promote transcription. The combinatorial nature of different histone marks therefore adds a layer of complexity in recruiting epigenetic modifiers and regulating cellular processes. For instance, Msk1/2-mediated H3S10 phosphorylation enhances binding of GCN5, which leads to acetylation of H3K14, methylation of H3K4, and inhibition of H3K9 methylation, the sequence of which results in open chromatin conformation [35]. Moreover, phosphorylation of H3S10 favors H3K9 acetylation since Aurora-B kinase can only bind unmodified or acetylated histone H3K9, thus preventing SUV39H1 binding and histone H3K9 methylation [36]. On the other hand, histone H3K9 methylation inhibits H3S10 phosphorylation and represses gene transcription [35]. Recently, phosphorylation of histone H3T6 by protein kinase C beta I was shown to be a major event in preventing LSD1 from demethylating histone H3K4 during androgen receptor-dependent gene activation [37].
Furthermore, this histone modification crosstalk can occur between different histones. Methylation of H3K4 and H3K79, which are involved in transcriptional activation, depend on, and are regulated by, H2BK123 ubiquitination [38]. The combination of specific histone modifications is due to the specificity of histone-modifying enzymes to a specific residue on their target substrate. Epigenetic marks on histone tails provide binding sites for specific domains of effector proteins [34]. For instance, bromodomains recognize and target acetylated residues, whereas chromo domains recognize methylation marks [39]. Together, the combinatorial and sequential modifications of histone tails provide a promising field of research that will allow for a better understanding of different cellular processes.
ROLE OF HISTONE MODIFICATIONS IN CELLULAR PROCESSES
Covalent modifications on histone tails are now established as key regulators of chromatin-based processes. Tis section discusses the role of different histone modifications in the regulation and coordination of transcription, DNA repair, and DNA replication (Fig. 5).
Transcription
In response to different stimuli, the regulation of gene expression in eukaryotes requires certain chromatin modifiers and specific histone modifications that allow for an "open", permissive chromatin (Fig. 3). These epigenetic modifiers facilitate and open the way for transcription factors to bind the DNA and activate a cascade of events resulting in gene transcription. On the contrary, other histone modifications and modifiers can result in transcriptional repression by inducing a condensed chromatin state and closing DNA accessibility to transcription factors. Thus, histone modifications dictate whether the chromatin state is transcriptionally permissive or not (Fig. 3).
Histone lysine acetylation has long been associated with transcriptional activation. Through decreasing histone charge, acetylation is believed to weaken histone-DNA interaction, relaxing the chromatin structure and opening the way for transcription machinery [40]. Moreover, acetylated histones may serve as docking sites for the recruitment of other transcriptional regulators that influence additional histone modifications [34]. Histone acetylation influences gene transcription at two levels; while global histone acetylation correlates with general transcriptional activity [41], specific promoter acetylation controls the activity of corresponding genes. It should be noted however, that specific promoter acetylation occurs in a context of global acetylation and deacetylation that regulates basal transcription levels to facilitate rapid transcriptional repression [42]. Reversal of acetylation is associated with transcriptional repression and the enzymes that carry out histone deacetylation are present in numerous repressive complexes [43].
As mentioned earlier, histone methylation plays two different roles in gene transcription. COMPASS-catalyzed histone H3K4 methylation is associated with RNA polymerase II in its initiating form. Methylated histone H3K36 is found at the 3' end of active genes in combination with the elongating form of RNA polymerase II [44]. Similar to the above mechanisms, methylated histone H3K4 may provide docking sites for downstream effectors that are involved with transcriptional activation, thus affecting gene expression [45]. Conversely, the methyltransferase for H3K9, H4K20, and H3K27 leads to a repressive effect on gene transcription. Chromatin modifiers that mediate these methylation marks function by inducing a repressive chromatin state and recruiting repressive complexes to transcription sites [45]. Lysine demethylation usually antagonizes the effect of methylation at the specific sites [46].
DNA repair
Eukaryotic cells continuously face numerous endogenous and exogenous genotoxic stresses that can cause deleterious DNA lesions, including DNA double-strand breaks (DSBs). To combat these threats, cells have evolved mechanisms of DNA damage repair to maintain genomic stability and prevent oncogenic transformation or development of disease [47]. Compacted chromatin can be a major obstacle in the orchestration of DNA repair and other chromatin-based processes. After the induction of DNA damage, chromatin must first be relaxed to give repair proteins access to the site of breaks. Biochemical and molecular studies have revealed the link between different histone modifications and DNA repair highlighting the major role of chromatin-remodeling enzymes in repair mechanisms [48]. For efficient repair, chromatin structure needs to be altered and access to the break sites must be available; both of which require post-translational histone modifications, adenosine triphosphate-dependent nucleosome mobilization, and exchange of histone variants. In this section, we focus on the role of post-translational histone modifications in DNA repair.
One of the earliest events in DSB signaling is the phosphorylation of H2AX, a variant of H2A. This phosphorylation is carried out by the phosphatase inositol-3 family of kinase and is spread over kilo-bases (in yeast) and mega-bases (in mammal cells) from the break site [49]. This modification is required for retention and accumulation of repair proteins to damaged sites [50]. Moreover, it has been shown that H2AX phosphorylation is required for the recruitment of HATs to break sites. The recruitment of HATs is mediated by Arp4 and leads to acetylation of the chromatin surrounding the breaks, thereby relaxing the chromatin and facilitating access for repair proteins [50]. Binding of NuA4 HAT complexes and the subsequent acetylation of H4 is concomitant with H2AX phosphorylation [51]. Moreover, defects in H3 acetylation results in sensitivity to DNA damaging agents, which is consistent with its importance in DNA repair [52]. Related to the important role of histone acetylation in DNA repair, a recent study provided evidence that TRRAP/TIP60 is essential for the recruitment and loading of repair proteins to the site of breaks [53].
The role of histone methylation in DNA repair has recently received considerable attention. Methylation of histone H4K20 in fission yeast was shown to be essential for the recruitment of Crb2, a checkpoint adaptor protein with homology to 53BP1, to sites of DNA breaks to insure proper checkpoint activation in response to DNA damage [54]. In human cells, 53BP1 may function in a very similar manner [55]. Interestingly, TIP60 binds to the heterochromatic histone mark H3K9me3, triggering acetylation and activation of DNA DSB repair. Although H3K9me3 is not required for the recruitment of TIP60 to sites of DNA damage, the interaction of TIP60/ATM with the MRN complex is sufficient for chromatin localization. However, the interaction with H3K9me3 is essential for TIP60 HAT stimulation and the initiation of downstream repair events [56].
DNA replication
DNA replication occurs during the S phase of the cell cycle and it is initiated at discrete sites on the chromosome called origins of replication. DNA replication is a delicate process for cells since it requires a high fidelity during the duplication of DNA sequences and maintenance and propagation of chromatin states. This cellular process involves several critical steps: access to DNA for the replication machinery, disruption of the parental nucleosomes ahead of the replication fork, nucleosome assembly on the daughter duplex of DNA, and propagation of the epigenetic state. All of these events are regulated by a network of histone-modifying complexes that control access to DNA and nucleosomal organization. Although the role of chromatin modifications in DNA replication remains poorly understood, several studies have provided evidence that histone post-translational modifications can control the efficiency and timing of replication origin activities [57]. As compacted chromatin can limit and prevent access for replication machinery to the DNA, it can be hypothesized that histone modifications play a critical role in setting the chromatin status for both early and late origins of DNA replication. Related to this idea, it has been shown in yeast that histone acetylation in the vicinity of the origin of replication affects replication timing. Indeed, higher levels of histone acetylation coincide with earlier induction of replication at an origin [57]. In humans, acetylation of histone tails has also been demonstrated to correlate with replication timing [58]. Consistent with the idea that acetylation opens the way for DNA replication machinery, several studies have shown that HAT HBO1 is associated with both replication factor, MCM2 and the origin recognition complex 1 subunit of the human initiator protein. These findings suggest that the targeting of histone acetylation to the origin of replication establishes a chromatin structure that is favorable for DNA replication [59]. Interestingly, ING5-containing HBO1 HAT complex associates with MCM2-7 helicase and appears to be essential for DNA replication in humans, which is consistent with the finding that depletion of either ING5 or HBO1 impairs S phase progression [60]. Recent studies in S. cerevisiae showed a dynamic regulation of acetylation of H3 and H4 around an origin of replication [61]. Further studies are needed to examine the exact mechanisms and implications of other histone modifications such as methylation, phosphorylation, and ubiquination. It is likely that histone phosphorylation, similar to acetylation, could also play roles in making DNA accessible to DNA replication machinery and in restoring chromatin to a compact configuration after DNA replication is completed.
EPIGENETIC ROLE OF HISTONE MODIFICATION IN GLIOMA
According to the nationwide, hospital-based cancer registry, as reported by the Korean Ministry of Health and Welfare in 2010, there were 10004 newly diagnosed brain tumors in a population of 49.9 million in 2010 [62]. Among them, most of the neuroectodermal tumors were gliomas (91.7%), which accounted for 15.1% of all primary brain tumors. Glioblastoma accounted for 5.2% of all primary tumors and 34.4% of all gliomas. Among histologically confirmed cases, glioblastoma accounted for 40.6% of all gliomas [62]. Despite recent advances in surgery, radiotherapy, and chemotherapy, survival of glioma patients remains poor. The 5-year survival rate of patients with low-grade gliomas is 30% to 70% depending on histology, and the median survival time is only 12 to 15 months for the most frequent malignant glioma, glioblastoma multiforme (GBM) [63].
Besides genetic alterations, epigenetic modifications are critical to the development and progression of cancer. The best-known epigenetic marker in gliomas is DNA methylation. Hypermethylation of the CpG island promoter can induce silencing of genes affecting the cell cycle, DNA repair, metabolism of carcinogens, cell-to-cell interaction, apoptosis, and angiogenesis, all of which may occur at different stages in glioma development and interact with genetic lesions [64]. In addition to DNA promoter hypermethylation, epigenetic alterations of histone modification patterns have the potential to affect the structure and integrity of the genome and disrupt normal patterns of gene expression, which may also contribute to carcinogenesis [13]. These modifications occur in different histone proteins, histone variants, and histone residues, involve different chemical groups, and have different degrees of methylation.
Alterations of histone modifications in glioma genesis
Mounted evidence from recent data shows that alterations at the histone level may also play a role in glioma genesis. These alterations encompass a globally deregulated expression of genes involved in histone modifications as well as changes in the histone modification pattern of individual genes (Table 4). Global aberrations at the histone level result from mutations in regulatory genes, as detected in a large-scale genomic analysis of GBM samples, including HDACs (HDAC2 and HDAC9), histone demethylases (JMJD1A and JMJD1B), and histone methyltransferases (SET7, SETD7, MLL3, and MLL4) [64]. Furthermore, altered expression levels of HDACs, due to reasons that have yet to be defined, have been linked to tumor recurrence and progression (HDAC1, HDAC2, and HDAC3) [65]. Histone modifications regulating individual genes have been reported in several studies. For example, a repressed expression of the tumor suppressor RRP22 and the cell cycle regulator p21, combined with an enhanced expression of the pro-proliferative transcription factor HOXA9 have been linked to alterations in histone modification patterns [66]. However, the actual functional roles of histone modifications in gliomas, and their potential to serve as biomarkers and/or therapeutic targets, still remain to be fully elucidated. Additionally, in order to determine the exact incidence and characteristic patterns of these alterations of histone modification in human gliomas, there is a necessity of further study and more analysis.
Alterations of histone modifications in GBM
As previously mentioned, epigenetically silenced loci, in addition to being hypermethylated DNA, are characterized by aberrant patterns of histone modifications. Silenced CpG island promoters are characterized by increased histone H3K9 methylation and loss of H3K9 acetylation. In embryonic stem (ES) cells, the dual presence of deactivating H3K27 methylation and activation-associated H3K4 methylation, called bivalent domains, is thought to create a "poised" chromatin state for developmentally regulated genes, allowing for silencing in ES cells and subsequent transcriptional activation or repression in differentiated cells [67]. Bivalent domains, along with additional repressive marks (dimethylated H3K9 and trimethylated H3K9), are found in embryonic carcinoma cells in genes that are frequently silenced by DNA hypermethylation in adult human cancer cells. These histone modifications are hypothesized to predispose tumor suppressor genes to DNA hypermethylation and heritable gene silencing [68].
There are many instances of genetic alterations and/or deregulated expression levels of genes encoding for histone-modifying enzymes. In acute leukemias for example, it is common to observe translocations involving the mixed lineage leukemia (MLL) gene, encoding for H3K4 methyltransferase [69]. These translocations result in MLL fusion proteins that have lost H3K4 methyltransferase activity. Mutations resulting in altered histone HAT activity also occur in cancer related diseases: CAMP response element (CRE) binding protein-binding protein mutations, abolishing HAT activity, cause Rubenstein-Taybi syndrome, a developmental disorder that is associated with a higher risk of cancer [70]. In GBM, there is also some preliminary evidence for the deregulation of genes controlling histone modifications. The gene encoding BMI-1, a member of the polycomb group complex that regulates histone H3K27 methylation, is frequently subjected to copy number alterations in both low- and high-grade gliomas, and BMI-1 deletions are associated with poor prognosis in patients [71]. It has also been reported that expression levels of some HDAC proteins are altered in GBM. Class II and class IV HDACs displayed decreased mRNA expression in GBMs compared to low-grade astrocytomas and normal brain samples, and overall, histone H3 was more acetylated in GBMs [72]. Large-scale sequencing of protein-coding genes in GBMs uncovered mutations in many genes involved in epigenetic regulation, including histone deacetylases HDAC2 and HDAC9, histone demethylases JMJD1A and JMJD1B, histone methyltransferases SET7, SETD7, MLL3, MLL4, and methyl-CpG binding domain protein 1 [73]. Screening of a large cohort of gliomas of various grades and histologies (n=784) showed H3F3A mutations to be specific to GBM and highly prevalent in children and young adults [74]. Furthermore, the presence of H3F3A/ATRX-DAXX/TP53 mutations was strongly associated with alternative lengthening of telomeres and specific gene expression profiles in GBM, which results explained the recurrent mutations in a regulatory histone [74]. These intriguing initial studies suggest that alterations in epigenetic mechanisms could be a major defect in GBM.
Global histone modification patterns as prognostic marker in glioma patients
Liu et al. [75] reported the relationship between multiple histone modifications and patient prognosis, which was analyzed by a recursive partitioning analysis (RPA), with progression-free survival (PFS) and overall survival (OS) as the primary end point. An RPA classification was carried out to test how histone modification might influence prognosis (Fig. 6). Patients with astrocytoma were classified into two separate groups based on acetylation of H3K9. Patients whose tumors expressed H3K9Ac in <88% of tumor cells (group 3) had a reduced survival rate compared with patients whose tumors had at least 88% of cells expressing H3K9Ac (group 2). In World Health Organization grade 3 tumors, patients whose tumors expressed H3K4diMe in <64% of tumor cells (group 6) had a reduced survival rate compared with patients whose tumors had at least 64% of cells expressing H3K4me2 (group 5). Acetylation of H3K18 also significantly influenced the survival of primary glioblastoma patients. Patients whose tumor expressed lower levels (<74% of tumor cells) of H3K18Ac (group 7 vs. group 8) experienced a greater survival rate. Meanwhile, trimethylation of H4K20 significantly influenced the survival of secondary glioblastoma patients; with a greater survival for patients whose tumor expressed higher levels (≥75% of tumor cells) of H4K20me3 (group 9 vs. group 10). Conclusively, these data suggest that the 10 groups defined the terminal classification of the 230 patients and were associated with significantly different PFS (p<0.0001) and OS (p<0.0001) (Table 5) [75].
THERAPEUTIC STRATEGIES TO OVERCOME HISTONE ALTERATION
Overview of potential epigenetic-based therapies for GBM
Epigenetic-based therapies, such as the DNMT inhibitor Decitabine (5-aza-2'-deoxycytidine) and the HDACi suberoylanilide hydroxamic acid (SAHA; vorinostat), are currently being tested in multiple cancers, although only HDACi is in trials for GBM. In contrast to genetic mutations, which are "hard-wired" once mutated, epigenetic mutations, such as promoter hypermethylation and histone acetylation status, are theoretically reversible through drug treatment or changes in the diet.
A major unresolved issue with epigenetic therapy for cancer is target specificity. First, some genes that require DNA methylation or histone deacetylation for silencing in normal cells could be unintentionally activated by agents that inhibit DNMTs or HDACs. Second, cancer genomes are characterized by both DNA hyper- and hypomethylation. Therefore, using drugs that reactivate silenced tumor suppressors may have the undesired effect of further activating oncogenes through hypomethylation. These problems should be addressed to gain a more complete understanding of the molecular events that may result from epigenetic-based therapy.
Histone deacetylase inhibitors for GBM treatment
HDACs catalyze the deacetylation of lysine residues within the N-terminal tails of core histones and non-histone proteins. As a result, their effects are complex and involve histone and non-histone substrates, and the mechanism of specificity for each HDAC is not well understood. In general, HDACs promote a closed chromatin structure that represses transcription. There are 18 known HDACs in humans, and they are divided into 5 main classes with different target specificities [76]. HDACs of Class I (HDACs 1, 2, 3, and 8), Class IIA (HDACs 4, 5, 7, and 9), Class IIB (HDACs 6 and 10), and Class IV (HDAC 11) all contain zinc in their active sites and are inhibited by HDAC inhibitors (HDACi) such as trichostatin A (TSA) and SAHA (vorinostat). Class III HDACs (sirtuins) do not contain zinc and are not inhibited by TSA or SAHA.
The rationale for using HDACi in cancer therapy is two-fold: first, HDACi promotes a more open chromatin conformation and might therefore permit better access for DNA-damaging agents to the chromatin, promoting apoptosis caused by these agents. Second, HDACi will reverse some of the aberrant epigenetic gene silencing in GBMs, presumably leading to cell-cycle arrest and apoptosis due to DNA damaging agents [77]. While it is not clear how consistently HDACi activates specific pathways from one GBM to the next, HDACi do synergize with DNA damaging agents in slowing the growth, or killing, glioma cells in vitro.
HDACi are comprised of several classes of compounds, including hydroxymates (SAHA, TSA), cyclic peptides (depsipeptide), aliphatic acids (valproic acid, butyrate), and benzamides. No single HDACi is effective against all HDACs. HDACi causes increased acetylation of histones and non-histone proteins and can reactivate p21, which contributes to cell-cycle arrest [78]. Non-cancerous cells are more resistant to the effects of HDACi, but the reasons for this selective sensitivity are unclear [79]. HDACi alters the expression levels of only a subset of expressed genes in transformed cells (~2-10%), and both increases and decreases in transcript levels have been observed [80].
SAHA is currently being tested as a monotherapy, and in combination with other therapies, in 5 phase I or I-II clinical trials for gliomas. SAHA targets Class I and II HDACs at micromolar concentrations, and preclinical studies found that it sensitizes glioma cells in vitro, ex vivo, and in vivo to chemotherapy and radiation [81]. SAHA treatment increased p21 promoter histone H3 acetylation in the U87 glioma cell line, and inhibited the proliferation of GL26 glioma cells implanted in mice [78]. Two of the current clinical trials are testing SAHA in combination with temozolomide; one also includes radiotherapy. A third trial consists of SAHA, isotretinoin and carboplatin, a fourth uses SAHA and bortezomib, and a fifth is testing SAHA as a monotherapy.
Trials are also currently underway for two additional HDACi, valproic acid (Depakene; Depakote) and depsipeptide (Romedepsin; FK-228), and there are additional compounds that remain to be tested in clinical trials. Valproic acid is being tested against GBM in combination with temozolomide in addition to radiation, and in a broader second trial against neuronal tumors and brain metastases in combination with etoposide. Depsipeptide monotherapy is being tested against high-grade gliomas in a study that is ongoing, but no longer recruiting participants. Valproic acid is known to be active against Class I and IIA HDACs at millimolar concentrations while depsipeptide is active against Class I HDACs at nanomolar concentrations. An additional HDACi not yet in clinical trials is pivaloyloxymethyl butyrate (AN-9), a derivative of butyrate. Butyrate is an aliphatic acid HDACi effective against Class I and IIA HDACs at millimolar concentrations. In addition, AN-9 shows efficacy in GBM cell culture and animal models [82]. AN-9 sensitized mouse GBM xenografts to radiation and showed decreased tumor growth and increased survival. There are several HDACi that have shown efficacy against cancer cells but have not yet been tested for gliomas, including Panobinostat (LBH589) [83] and Belinostat (PXD101) [84]. The discovery and development of new epigenetic enzyme-targeting compounds is an area of active research in the pharmaceutical industry.
FUTURE DIRECTIONS FOR EPIGENETIC HISTONE MODIFICATION IN GBM
Epigenetic studies of GBM are poised to 1) make substantial contributions to the understanding of GBM biology, 2) identify new predictive biomarkers, and 3) discover novel targets for therapy. New models, such as GBM patient-derived tumor stem cells grown in neurosphere culture, may be a valuable addition to epigenetic research into GBM, particularly if the epigenetic profiles of the corresponding primary tumors are retained, as has been shown in gene expression patterns and invasive growth patterns of these cells [85]. Epigenomic profiling of DNA methylation, histone modifications and non-coding RNAs (such as microRNAs) in primary tumors, orthotopic xenografts, and tumor neurospheres are strategies that will likely uncover many additional epigenetic alterations in GBMs, and potential targets for therapy.
There are still many questions remaining about the role of epigenetics in GBM. The causes and consequences of epigenetic alterations are still mostly unknown, and the relative contributions of genetic and environmental factors to epigenetic alterations have not been quantified. It is still unclear as to why some genes or pathways are more affected by epigenetic alterations than they are by genetic alterations (or vice versa). It is clear, however, that simultaneously examining both genetic and epigenetic defects, complemented with functional studies, will be essential in answering these questions. It will also be important to understand the effects of HDACi on the entire cancer acetylome to elucidate the molecular consequences of this treatment strategy.
Therapies that combine both DNMT and HDAC inhibitors may be an effective strategy against GBM. A dual treatment approach may have a synergistic effect on gene activation, and could allow lower doses of each drug to be used. Such a strategy is being tested in a clinical trial for myelodysplastic syndrome and acute myelogenous leukemia using the DNMT inhibitor Decitabine with or without valproic acid (clinicaltrials.gov ID NCT00414310).
An area that is mostly unexplored in GBM is the development and testing of drugs directed against histone modifications other than acetylation. H3K27 methylation at promoter regions of silenced tumor suppressors could be targeted to reactivate these genes by, for example, using the S-adenosylhomocysteine hydrolase inhibitor 3-Deazaneplanocin A (DZNep) [86]. However, it should be noted that the degree of specificity of DZNep for inhibiting H3K27me3 has not yet been fully determined because of its strong toxicity. As epigenetic epigenetic modifications are better understood and more types are discovered, additional epigenetic drug targets can be tested in GBMs and other cancers.







 XML Download
XML Download