

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Tumor replication is associated with two types of genes. One is the oncogene and the other is the tumor suppressor gene. The oncogene is activated by amplification, translocation and point mutation. The function of the tumor suppressor gene is inactivated by point mutation, deletion, and cytosine-guanine dinucleotides (CpG) island hypermethylation.
Scientists have studied genetic and epigenetic alteration; which one plays a larger role in tumor development. In many studies, it turns out that key regulating genes have tumor suppressor genes silenced by developing epigenetic changes [1]. Among them, CpG island methylation is being actively studied. There are many reports on the relationship between hypermethylation of tumor suppressor genes and malignant tumors in extraneural tissue. Aberrant methylation of 5' CpG islands is thought to play an important role in the inactivation of tumor suppressor genes in several types of cancers [2]. Epigenetic silencing associated with aberrant methylation of the promoter region CpG island is one of mechanisms, leading to the loss of tumor suppressor function in human cancers [3]. Published reports have shown that in addition to the frequent amplification and deletion of the epithelial growth factor receptor gene, the main genetic events affecting tumor suppressor genes such as O-6 methyl guanine DNA methyltransferase (MGMT), the members of the inhibitor of cyclin-dependent kinase 4 A initiated cell-cycle arrest pathway p14, phosphatase and tensin homolog, p16, tumor protein 53, retinoblastoma protein, and protocadherin-gamma-A11, paternally-expressed gene 3, distinct subgroup of the ras family member 3, epithelial membrane protein 3, and caspase-8, are commonly aberrantly methylated in malignant brain tumors [4,5,6,7,8]. Ras association domain family member 1 (RASSF1A) has been shown to be downregulated by methylation in cancer, and MGMT is located in areas commonly deleted in astrocytomas. Specific changes in methylation associated with different subtypes of gliomas also have been reported. However, these studies were limited to only a few samples or a few genes, or to only limited subtypes of gliomas [9]. Profiling CpG islands methylation indicates that some genes are more frequently methylated than others, and each tumor type is associated with a unique set of methylated genes [10,11].
This study was performed to investigate whether the promoter hypermethylation of cancer-related genes is involved in the development of malignant gliomas. For this purpose, the authors correlated methylation and expression levels to identify genes that are transcriptionally regulated by epigenetic alterations.
MATERIALS AND METHODS
Patients and specimens
Tissue samples of 29 consecutive patients with pathologically confirmed malignant gliomas were included in the present study. The number of male patients was 18, and 11 females. Various malignant brain tumors contained 15 cases of glioblastomas (GBMs), 8 cases of anaplastic astrocytomas, and 6 cases of anaplastic oligodendrogliomas. All patients received neurosurgery for primary tumors at our institution between January 2000 and December 2006. The tumor specimens obtained in the operation room were fixed in formalin for 24 hours, paraffin-embedded and used for later analysis. Clinical information was obtained from the patients' charts. Peripheral blood was analyzed as controls. Permission to perform this study was provided by our local Institutional Review Board. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
DNA extraction
Twenty nine tissue samples gained through an operation of malignant brain tumors were put in a 2 mL container, then cancer cells were homogenized by a tissue crusher. Gained through this process, tumor tissue was limited to cases involving over 90% of cancer cells. Fixed by formalin, paraffin-embedded samples of malignant brain tumors were cut into 5 to 6 slices to a thickness of 7 mm. After paraffin removal by xylene and alcohol treatment, deoxyribonucleic acid (DNA) was extracted following the manufacturer's instructions. For extraction of DNA from blood, 10 mL blood and ice-cold lysis buffer [1.28 M sucrose, 20 mM Tris (pH 7.5), 10 mM MgCl2 2% Triton X-100] was poured into a 50 mL container and mixed. This solution was put into ice for 10 minutes then centrifugated for 15 minutes, and the upper portion was thrown away. After 700 µL TE9 solution (10 mM Tris-hydrogen chloride, 1 mM ethylene diamine tetra acetic acid, pH 8.9) was added into the container, it was cultured and floated.
Next, the mixture of 70 µL sodium dodecyl sulfate and proteinase K was made to react for from 48 to 60 hours in 60℃. Then additional mixture of 10 µL sodium dodecyl sulfate and proteinase K was added and made to react for from 24 to 48 hours in 60℃. After that, this solution was put into the boiled water for 10 minutes to inactivate proteinase K and cooled at room temperature. Then the same amount of phenol/chloroform/isoamyl alcohol was put, mixed and centrifugated. Next, the upper side of the solution was moved to another container, and 3 M sodium acetate (pH 5.2) was added, a tenth of that solution, and twice amount of absolute alcohol were added to get the DNA spool. After cleaning with 70% alcohol and dried in the air, the DNA spool was dissolved in 500 µL LOTE (3 mM Tris, 0.2 mM ethylene diamine tetra acetic acid, pH 7.5), then the absorbance was measured and kept at 4℃ until it used.
Methylation of CpG island of DNA promoter
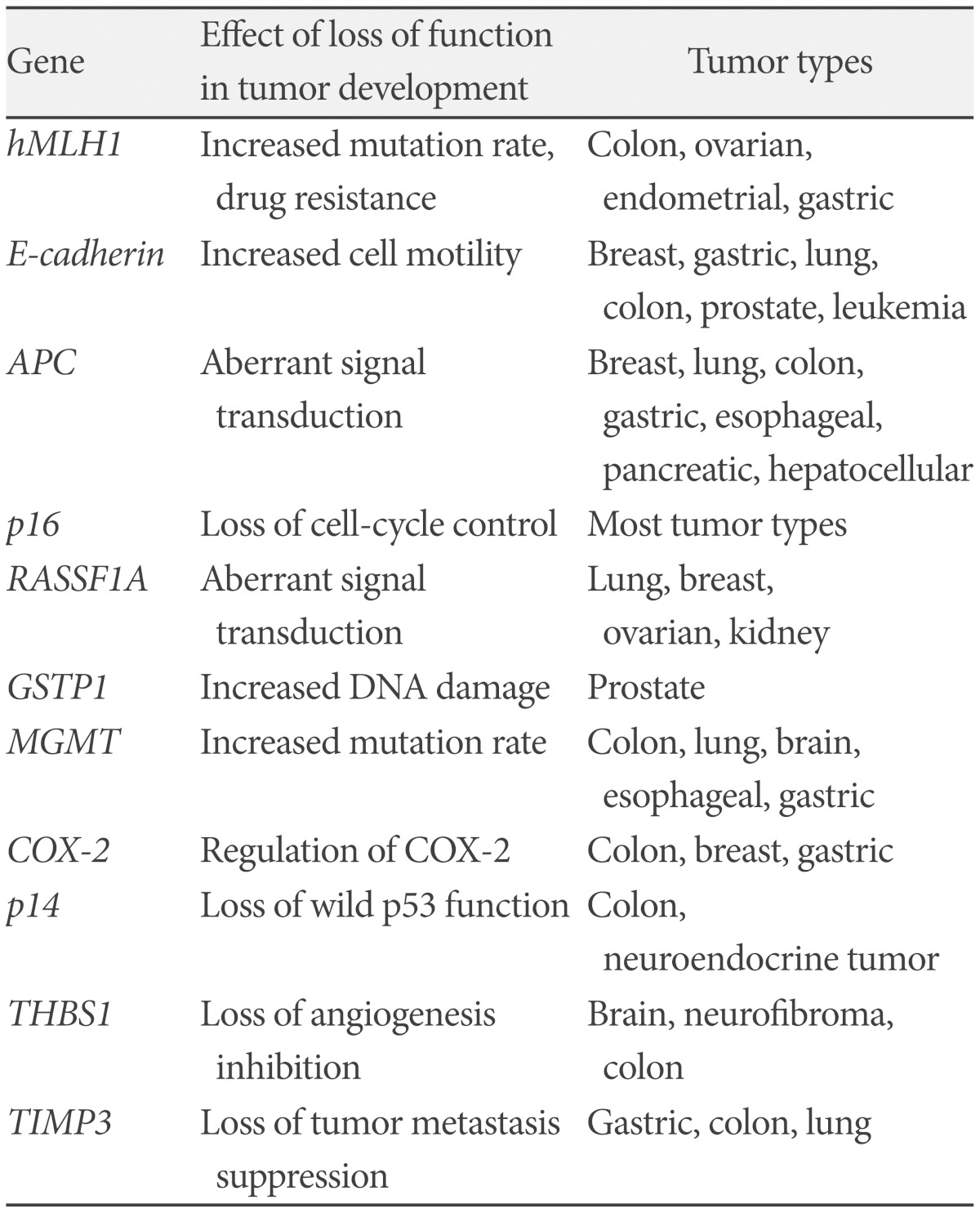
In this study, the degree of methylation of the promoter of 11 tumor-related genes was analyzed (Fig. 1). These 11 genes were selected for the known involvement in carcinogenesis and frequent epigenetic inactivation in malignant brain tumors or other functions in other cancers; cell cycle regulation: p14, p16, cyclooxygenase 2, DNA repair or protection: human mutL homologue 1, MGMT, glutathione S-transferase pi 1 (GSTP1), signal transduction: adenomatous polyposis coli (APC), RASSF1A, angiogenesis: thrombospondin 1 (THBS1), tissue inhibitor metalloproteinase 3, metastasis and invasion: epithelial cadherin (E-cadherin). Among these, p16 and RASSF1A genes are commonly methylated in a variety of cancers. But other genes are methylated in specific cancers. For example, GSTP1 is methylated in more than 90% of prostatic cancer and APC is methylated in 90% of esophageal cancer (Table 1) [12,13].
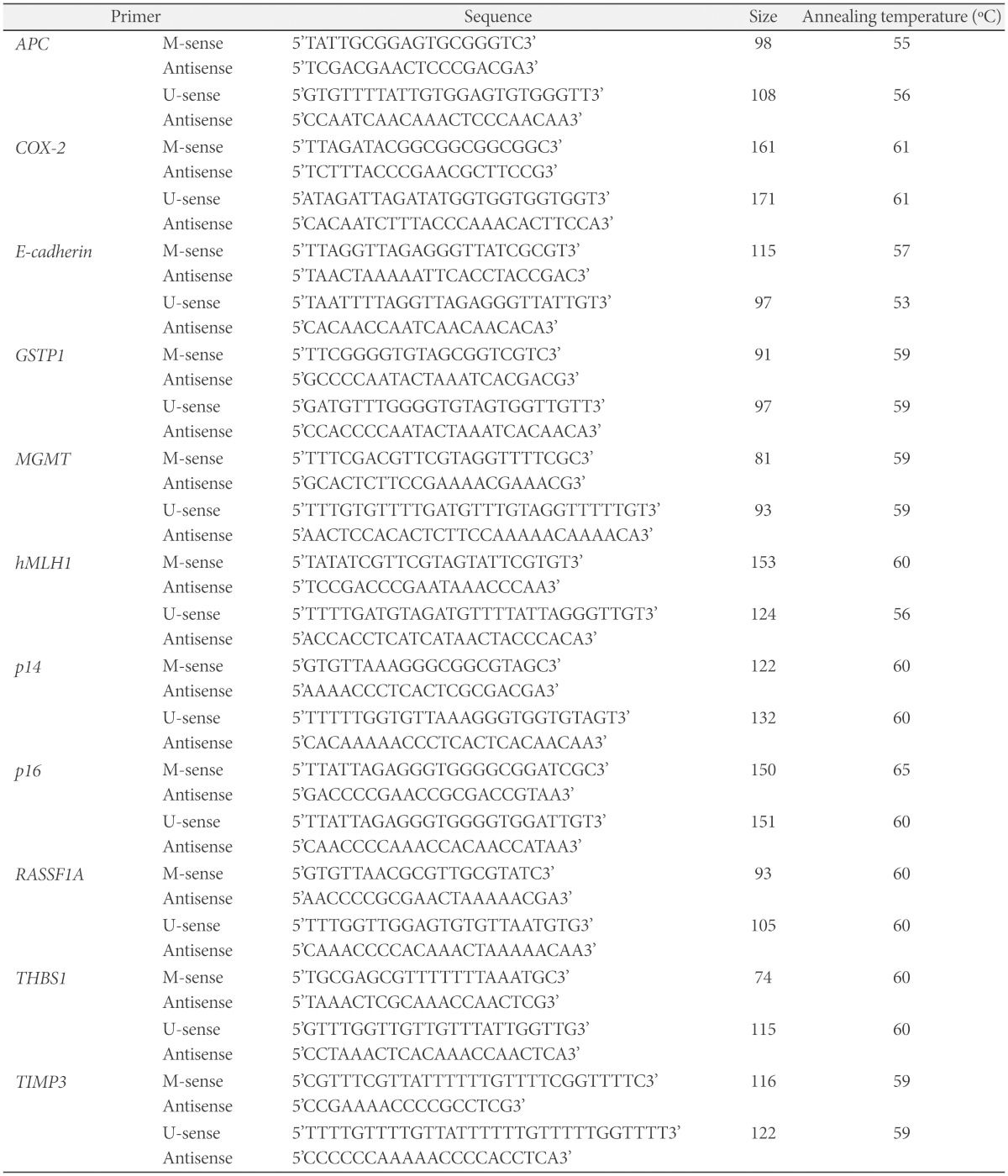
To analyze the methylated condition of a gene, a method of methylation-specific polymerase chain reaction (PCR) reported by Clark et al. [14] and Herman et al. [15] was used. The CpG island position of methylcytosine was measured by using the Clark and Herman method. Cytosine methylated by bisulfite modification was not changed, and unmethylated cytosine was changed to uracil. It was checked whether cytosine was methylated or not through the amplification of PCR. First, extracted DNA was made to react with sodium bisulfite, hydroquinone and sodium hydroxide, and then deamination reaction was induced to change the unmethylated cytosine into uracil. Next, DNA was extracted by using the Genomic DNA purification kit (Wizard®; Promega, Madison, WI, USA), and desulfonation induced with sodium hydroxide. Then it was settled with ethanol and amplified using PCR. To amplify DNA unmethylated by CpG island, the unmethylated primer set of each gene was used, and the methylated primer set of each gene was used to amplify DNA methylated by CpG island of DNA promoter. The size of each gene was measured by base pair. Each base sequence of primers was in the following Table 2. The PCR reaction was 20 µL reaction, adding deoxy-nucleotide tri-phosphate 250 mM, 10 pM primer each, DNA 150 ng, MgCl2 1.25 mM and Taq polymerase 0.5 U to 1×PCR buffer solution. The solution was made to react in the condition of a temperature of 95℃ fo r 15 minutes, and for 50 seconds at various annealing temperature depending on each primer. Then it was kept to react at 72℃ for 50 seconds and repeated 40 times. This process finally finished after additional 10 minutes at 72℃. The PCR products, the result of the reaction, were visualized in 4% Agarose gel (MetaPhor®; Lonza, Rockland, NY, USA), and each base pair could be confirmed.
RESULTS
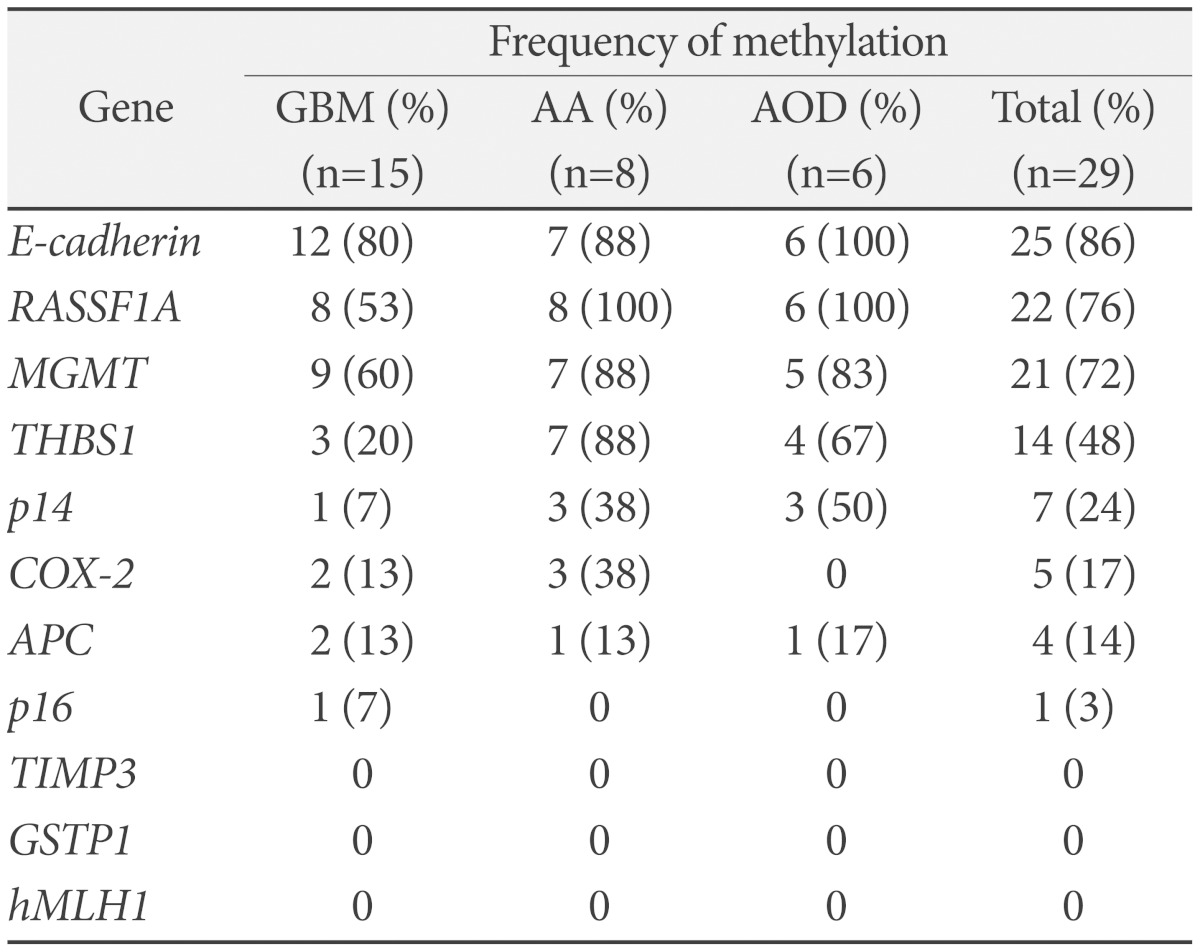
All of 29 malignant gliomas were the promoter of methylation of at least two genes per sample. Methylated genes range from one to six. Of 29 tumors, 28 (97%) showed concurrent hypermethylation of three or more genes.
In GBMs, eight genes were methylated at frequencies ranging from 7 to 80%. E-cadherin methylated in 12 of 15 samples, MGMT in 9 cases, RASSF1A in eight cases. In anaplastic astrocytomas, seven genes were methylated at frequencies ranging from 13 to 100%. RASSF1A methylated in all cases of eight samples. E-cadherin, MGMT and THBS1 methylated in seven cases. In anaplastic oligodedroglioma, six genes were methylated at frequencies ranging from 17 to 100%. RASSF1A and E-cadherin methylated in all cases of six samples, MGMT in five cases, and THBS1 in four cases (Table 3).
DISCUSSION
Epigenetics, which is not the consequence of a change in DNA sequence, is brought about by three systems, which include include DNA methylation, histone modification and ribonucleic acid-mediated action [16]. Tailoring the correct anticancer drug to the right cancer patient should be one of the foremost goals of oncologic treatment. Aberrant DNA methylation is a common hallmark of human cancer, and so the methylation-associated silencing of certain genes is another good candidate in the pharmacogenomics field [17].
CpG island, the size of 0.5-2 kilobase and a short stretch of DNA in which the frequency of the CG sequence, is concentrated and located in the promoter which controls the transcription of genes [18,19]. CpG island is a useful marker for genes in organisms containing 5-methylcytosine in their genomes [20]. In addition, methylation of cytosine inhibits the transcription, and consequently leads to the inactivation of genes. It is also reported in the process of the inactivation of X chromosome and imprinted gene [21]. The case of methylation of normal genes is an exceptional result, and CpG island is not always methylated in most genes involving tumor suppressor genes. The function of tumor suppressor gene is inactivated when a hypermethylation happens from some causes, which leads to the development of several tumors [22].
DNA methylation is an epigenetic event. There is no change in the gene sequence, but some changes of gene activity and methylation at the gene promoter region, and consequently the expression of genes is reduced. It is also reported that the epigenetic change, especially the hypermethylation of the DNA promoter gene plays important roles in this process. The methylation of the CpG island located in the promoter of tumor suppressor genes inhibits the transcription of genes, which loses the expression of proteins. These days the tumor with hypermethylation of many genes at the same time has been named as the CpG island methylation phenotype.
The present study shows that the Bead array technology (GoldenGate®, Illumina, San Diego, CA, USA) is a powerful method for classification purposes, allowing the identification of specific methylation profiles related to different ways of tumorigenesis. However, due to the heterogeneity of methylation, the sensibility and specificity of methylation arrays can be variable among the genes. Therefore, as demonstrated for the assessment of MGMT methylation, results obtained on individual genes need to be validated using another technique [6]. The large number of genes that undergo hypermethylation in a wide variety of cancer types suggests a key role for changes in DNA methylation during the initiation and progression of cancer. Promoter hypermethylation and silencing of the associated genes, especially MGMT is widespread in GBM. Many of the genes that are targeted by the methylation machinery in cells play a role in regulating apoptosis and important cellular functions such as cell growth, differentiation, and DNA repair. The most significant gene that has been described is MGMT, which is methylated at high frequencies in malignant gliomas [23].
Our result showed that 100% of malignant gliomas exhibited the hypermethylation of the promoter at least one of tested genes, and none of them is hypermethylated in peripheral blood. In at least one of the genes, 28 (96.55%) showed that hypermethylation of 3 or more genes. And RASSF1A, E-cadherin, and MGMT are frequently methylated in neuroepithelial tumors such as glioblastomas, anaplastic astrocytomas, and anaplastic oligodendrogliomas [24,25]. So, aberrant hypermethylation profile is associated with malignant gliomas, and it means epigenetic change plays a very important role in the malignant changes of brain tumors.
The limitation included in this study is based on the heterogeneous group of the tumor samples. Twenty nine malignant brain tumors were studied, and these samples were divided into three groups. The number of each group was not the same. Also, this study is only based on a single center study. Last, this study involves only a small number of patients and has no statistically meaningful relationship as to which gene is related with malignant brain tumors. Not every single patient's prognosis ccould be checked, specifically long time survival, so clinical usefulness of methylated tumor related genes cannot be assessed. This study is a preliminary investigation that shows whether promoter hypermethylation of cancer-related genes is involved in the development of brain tumors. Therefore, multi-center study and more patients for further evaluation regarding hypermethylated cancer-related genes except those already known will be needed. Also, future studies pertaining to low grade gliomas will be needed.
Conclusion
Aberrant hypermethylation of CpG islands may be involved in malignant change of brain tumors. The identification of CpG promoter methylation status by using methylation-specific PCR technology allows the selection of patients most likely to benefit from chemotherapy. So, multi-center study and more patients for further evaluation about hypermethylated cancer-related genes except already known will be needed.

 XML Download
XML Download