

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Atypical teratoid rhabdoid tumor (ATRT) is a highly malignant brain tumor which is mainly found in pediatric patients, especially younger than 3 years old, with poor prognosis [1]. Histologically, ATRT is known that they composed of diffuse proliferation of atypical large cells showing eccentrically located nuclei and abundant eosinophilic cytoplasm (rhabdoid features) with prominent nucleoli. But this microscopic characteristic alone is often insufficient to differentiate an ATRT from other malignant brain tumors [2]. It is well known that ATRT is associated with deletion or mutation of INI1 (hSNF5/SMARCB1), a member of the SWItch/Sucrose NonFermentable chromatin remodeling complex located on chromosome 22q11 [2,3,4]. Now, detection of the loss of INI1 protein expression is a diagnostic hallmark of ATRT and malignant rhabdoid tumors (MRT) [5]. ATRT in adult patients have been rarely reported, but their prognosis is quite better than that of a pediatric group [3,6,7,8]. They are mainly located in the cerebral hemispheres, and ATRT in posterior fossa and spinal cord is not uncommon. Recently, it has been reported that sellar or suprasellar region is also preferential site for ATRT [9,10,11,12,13]. Herein, we report a rare case of a sellar and suprasellar ATRT in a female adult patient who was successfully treated with a multimodal approach.
Go to : 
CASE REPORT
History and examination
A 42-year-old female patient presented with progressive visual disturbance. She did not have any other medical history before but hypertension and pulmonary tuberculosis. Humphrey test revealed defect of more than three quarters of her bilateral visual field. No other neurologic deficits were found. Preoperative magnetic resonance imaging (MRI) showed a heterogeneously enhanced solid and cystic mass involving the sella and suprasellar region with compression of an optic apparatus (Fig. 1). The solid part of the tumor showed isointense signal with heterogeneous enhancement on T1-weighted images and slightly hyperintensity on T2-weighted images.
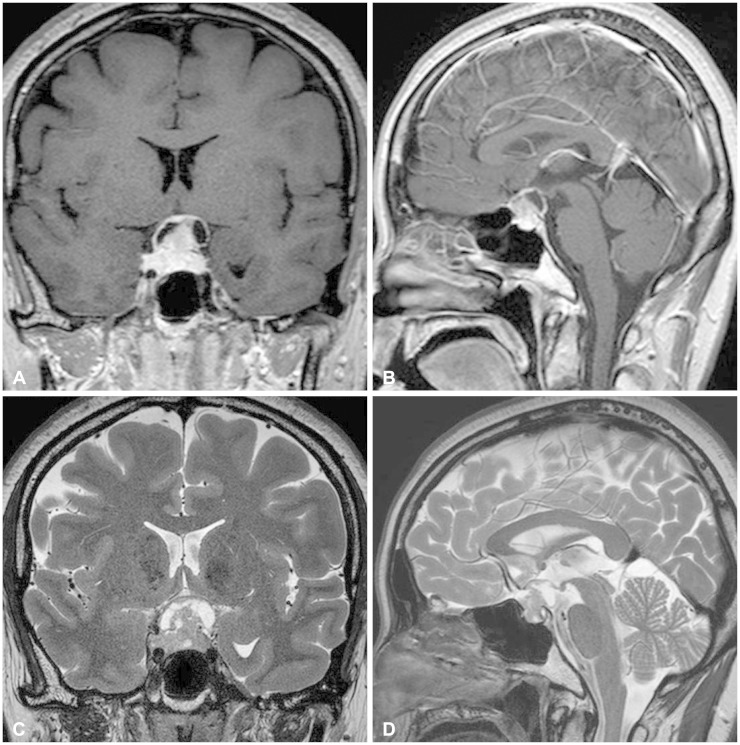 | Fig. 1Preoperative magnetic resonance imaging shows a heterogeneously enhanced mass involving the sella in a T1-weighted gadolinium-enhanced coronal (A) and sagittal view (B). T2-weighted coronal (C) and sagittal (D) images reveals solid and cystic components of the tumor with compression of an optic apparatus.
|
Operation
The tumors were approached with a transsphenoidal route. A yellowish-grey mass was seen over a pituitary gland and no definite diaphragm was identified. The tumor was not dissected from a pituitary gland so that a part of a pituitary gland was resected together with the tumor mass. The frozen section of the tumor showed the presence of high grade malignant tumor cells and not a benign tumor such as a pituitary adenoma. Most of solid part of the tumor was removed, however, and a part of the thin tumor capsule was left as it was very adhesive to the optic nerves and chiasm. Intraoperative cerebrospinal fluid (CSF) leakage was noted and sellar floor reconstruction was done with our conventional method [14].
Pathological findings
On low magnification, hematoxylin-eosin stain revealed relatively homogenous population of tumor cells with very high cellularity. At the inferior margin of the tumor mass, it showed a continuation between the tumor mass and pituitary gland. Higher magnification view demonstrated a few tumor cells showing typical rhabdoid features with eccentric nuclei and prominent nucleoli (Fig. 2). The loss of INI1 was confirmed by immunohistochemistry (Fig. 3), which was consistent with ATRT. Immunohistochemical stains for glial fibrillary acidic protein, CD3, CD43, CD45, CD56, CD138, CD20, synaptophysin, and cytokeratin were all negative except CD99.
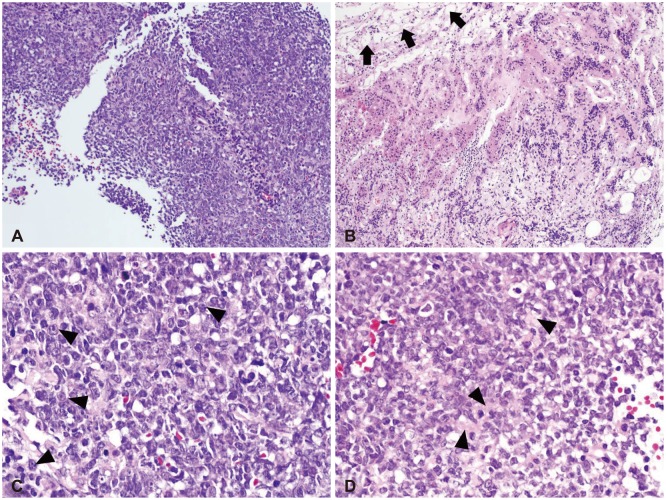 | Fig. 2Low power views show relatively homogenous population of tumor cells with very high cellularity (A). At the inferior margin of the tumor mass, pituitary glandular structure (black arrows) is seen (B). High power views demonstrate a few tumor cells showing typical rhabdoid features with eccentric nuclei and prominent nucleoli (black arrowheads) (C). Abundant eosinophilic cytoplasm (black arrowheads) is noted (D). H&E, original magnification ×100 (A and B), ×400 (C and D).
|
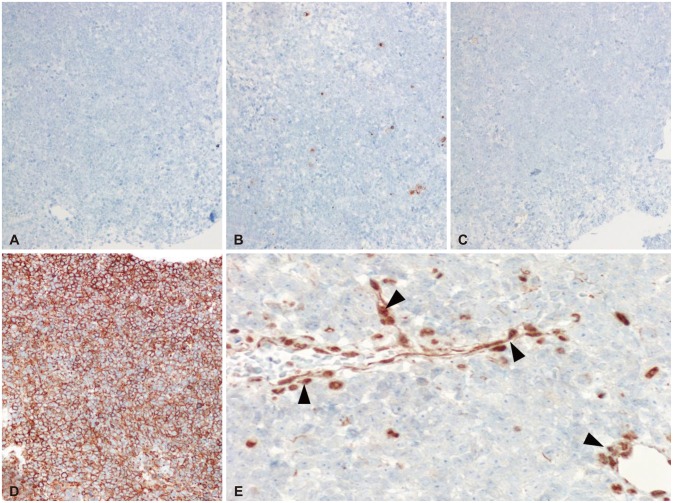 | Fig. 3Immunohistochemistry (IHC) was negative for CD20 (A), cytokeratin (B), synaptophysin (C), and strong positive for CD99 (D). IHC stain for INI1 is negative in tumor cells whereas it is positive for endothelial cells (black arrowheads) (E). CD20-IHC, original magnification ×200 (A), cytokeratin-IHC, ×200 (B), synaptophysin-IHC, ×200 (C), CD99-IHC, ×200 (D), and INI1-IHC, ×400 (E).
|
Postoperative course and further treatments
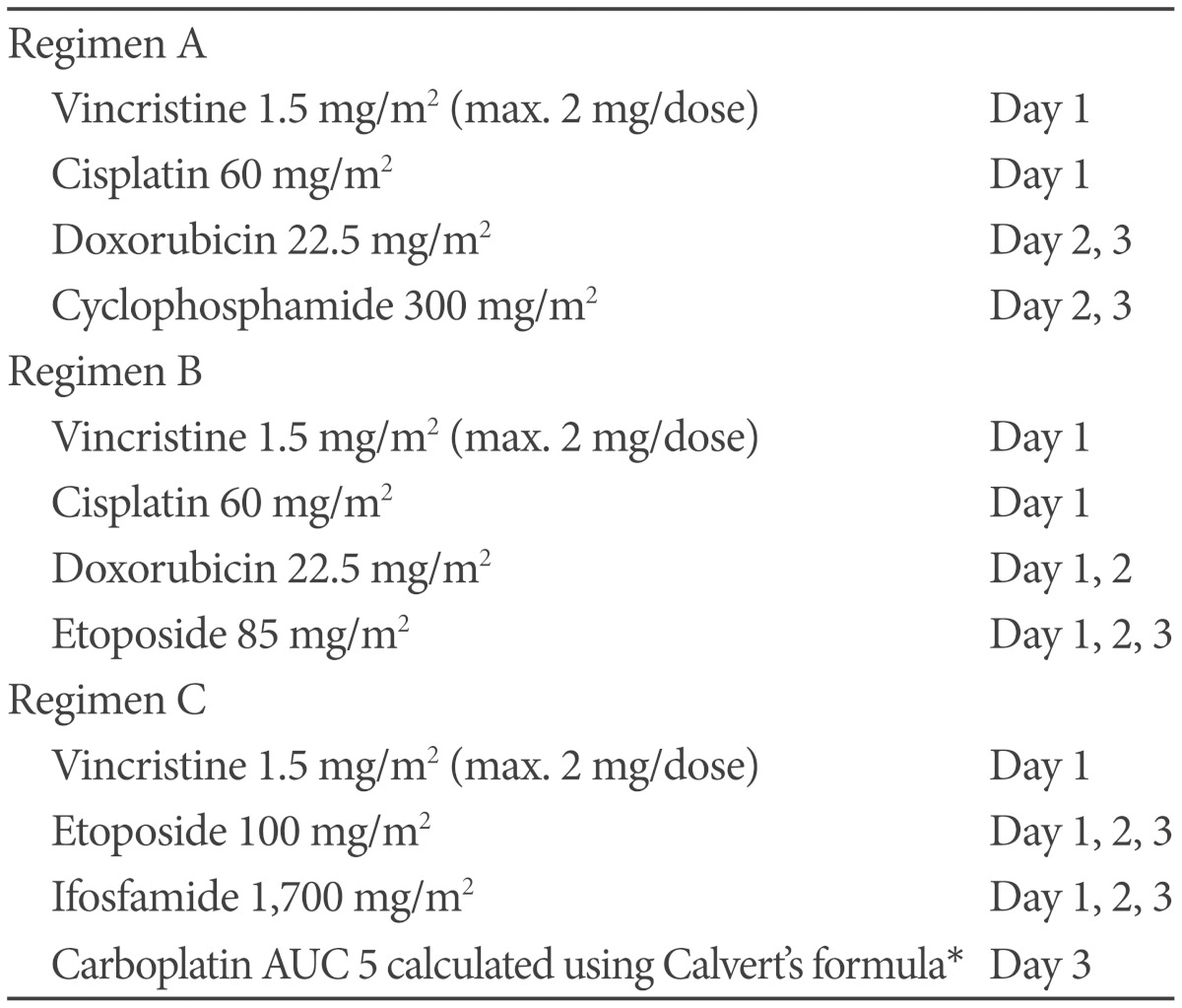
Based on Humphrey test, patient's visual field defect was much improved and her visual acuity was also improved. Cytology of CSF and spinal MRI were negative of tumor. At 27 days after surgery, she received radiotherapy that consisted of craniospinal irradiation 3,600 cGy, including spinal irradiation with proton beam therapy, and local boost 1,800 cGy, with 30 mg/m2 each week of intravenous cisplatin as radiosensitizer during radiotherapy. Subsequently, she received 11 courses of multiagent chemotherapy for 10 months that consisted of vincristine, cisplatin, adriamycin, etoposide or cyclophosphamide, alternating with vincristine, etoposide, ifosfamide and carboplatin. This regimen was modified from previously known regimens (Table 1). Prophylactic granulocyte colony stimulating factor was given during myelosuppression. She completed chemotherapy without any significant toxicity and was followed at the outpatient clinic for more than 2 years with no evidence of recurrence or other complications (Fig. 4).
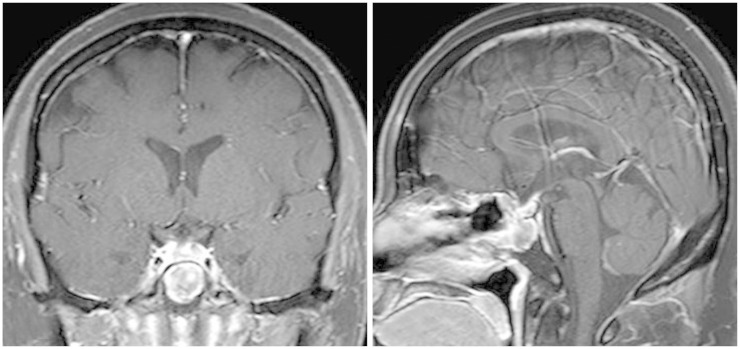
Go to :
DISCUSSION
Atypical teratoid rhabdoid tumors are rare malignant brain tumors usually diagnosed in children younger than 3 years old [1]. Their clinical outcomes are generally very poor despite aggressive treatment. ATRTs are polyphenotypic tumors as they show various pathological features with various immunohistochemistry, which is very similar to the renal or extrarenal MRT [4]. Alteration of the hSNF5/INI1 gene located on chromosome 22q11.2 is the most distinctive feature of an ATRT, MRT and epithelioid sarcoma [5,15,16]. Lack of INI1 can be mediated by deletion of chromosomes 22q11.2, mutation of the INI1 gene, and loss of expression of the INI1 protein [12,15,17,18]. The loss of the INI1 expression is now regarded as a pathognomonic finding of an ATRT [2,4,19]. Adult-onset ATRTs are very rare. Up to date, 31 cases of adult ATRTs have been reported in a literature [20,21]. Most of adult-onset ATRTs are located in the supratentorial area, whereas about 50% of ATRTs are found in this area [21,22]. Very distinguished from pediatric ATRTs, adult-onset ATRTs have much more favorable clinical course as many of reported cases showed long term survival [21]. Not only different biological nature, but also the use of stronger radiation might be the causes of big difference in their prognosis [21].
Recently, it has been reported that sellar and suprasellar region is a preferential site of adult ATRTs, where no ATRT in this site has been found in pediatric population [9,10,11,12,13]. Only seven cases of adult-onset ATRTs in sellar and suprasellar region have been reported in the literature so far (Table 2) and our case is the 8th. Most of the cases presented visual loss or limitation of ocular movement caused by sellar tumors that invaded suprasellar space and cavernous sinus. Their diagnosis was confirmed by the loss of INI1 by fluorescence in situ hybridization and immunohistochemistry. Considering ATRT is known to have a slight male predilection [22], it is very interesting that all of 8 cases were female patients.
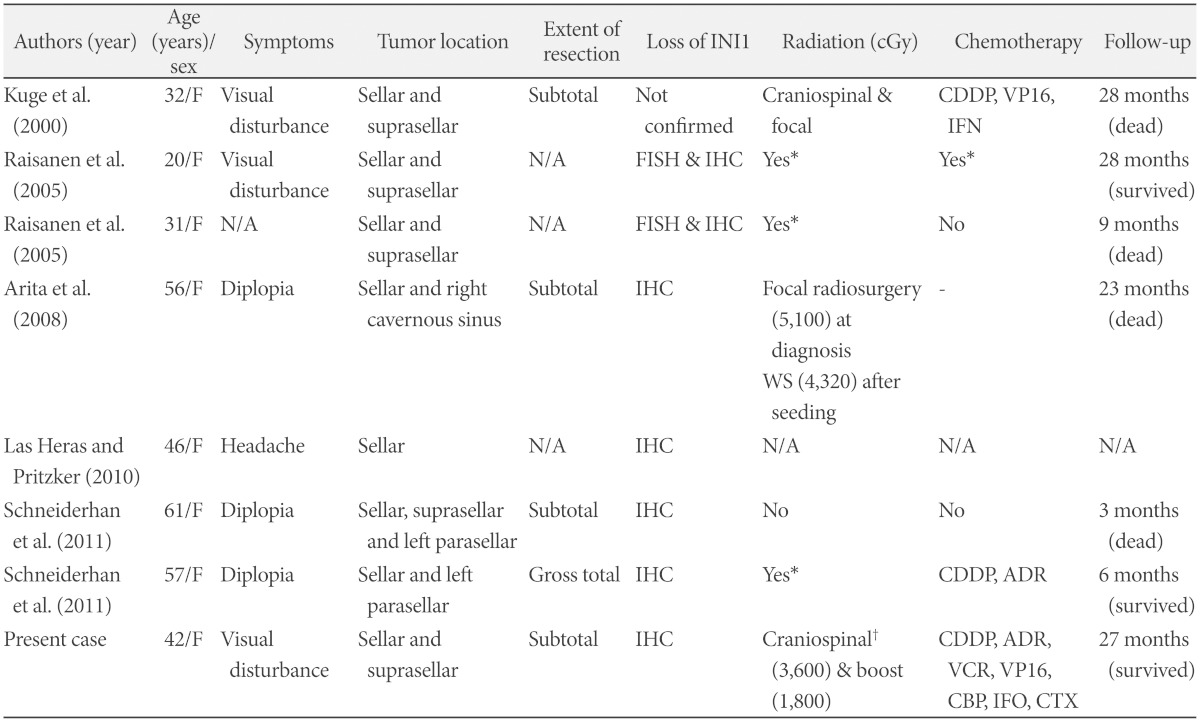
Table 2
Summary of adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor in the literature
*area of radiation or chemotherapeutic regimen were not specified, †spinal irradiation was given with protons. ADR: doxorubicin, CBP: carboplatin, CDDP: cisplatin, CTX: cyclophosphamide, FISH: fluorescence in situ hybridization, IFN: interferon, IFO: ifosfamide, IHC: immunohistochemistry, N/A: not assessed, VCR: vincristine, VP16: etoposide, WS: whole spine
![]()
There is no established treatment strategy for adult ATRT. It is well known that surgery (total resection) and radiotherapy are the mainstays in the treatment of ATRT [23]. In childhood ATRT, many recent studies used high dose chemotherapy and autologous stem cell rescue, with deferred radiotherapy, because of the very young onset age and long-term effect of radiotherapy [23,24]. However, treatment strategies of adult ATRT may be different from that of pediatric ATRT. We treated her with radiotherapy including craniospinal irradiation of 3,600 cGy and concurrent cisplatin as radiosensitizer, with consideration of a high rate of local relapse and possibility of leptomeningeal seeding.
Another issue is whether adjuvant systemic chemotherapy is beneficial. Four out of 6 cases, of whom treatment information were available, were treated with combined treatment modalities including surgery, chemotherapy, and radiotherapy. Three of these patients survived longer than 24 months (Table 2). In contrast, 2 patients treated with only surgery and irradiation died of tumor progression, suggesting the beneficial effect of chemotherapy. The optimal chemotherapeutic regimen for treatment of ATRT is still unknown. In previous reports, many kinds of chemotherapeutic agents were used in childhood ATRT without definite successful outcome [20,23,24,25]. Recently, Chi et al. [25] reported promising results (2-year progression-free survival of 53±13%) after treatment of childhood ATRT with upfront radiotherapy and multidrug chemotherapy including intrathecal and intraventricular chemotherapy without high-dose chemotherapy. In addition, ifosfamide, carboplatin, and etoposide is one of the most widely used regimens for the treatment of adult ATRT [20]. Based on these results, we combined these two regimens with dose modifications for adults (Table 1). We were concerned about myelotoxicity of craniospinal irradiation and chemotherapy, so proton beam radiation to spine was given to preserve of marrow function and GCSF was administered prophylactically. Therefore, our patients finished chemotherapy without serious infections and adverse events.
We reported a case of sellar and suprasellar ATRT in an adult female who was successfully treated with aggressive multimodality treatment. Despite its rarity, ATRTs should be considered in the differential diagnosis of an unclear malignant sellar or suprasellar lesion in adult patients. Furthermore, the optimal treatment strategies for adult ATRT patients could be established.
Go to :
 XML Download
XML Download