

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
High-grade gliomas such as anaplastic astrocytoma [World Health Organization (WHO) grade III] and glioblastoma multiforme (GBM, WHO grade IV) are the most common and aggressive brain tumors in adults. Composed of a heterogeneous mixture of poorly differentiated neoplastic astrocytes, high-grade glioma preferentially occurs in the cerebral hemispheres. Despite intensive basic and clinical studies and the development of various therapeutic modalities, the average survival of patients with GBM post-diagnosis remains less than 15 months, and most GBM patients eventually experience tumor relapse and outgrowth within 7 months of initial radiation therapy [1-4]. Thus, owing to its poor prognosis and difficult management, development and translation of new anti-glioma therapeutic approaches into the clinic are urgently needed.
Identifying the cellular origin of glioma provides the opportunity to improve the understanding of this disease. Although oncogenic transformation of differentiated glial cells is generally thought to represent glioma initiation, this hypothesis has not been thoroughly investigated through basic or clinical studies. Moreover, although astrocytoma possesses some morphological features of cells that are similar to those of mature astrocytes, the origin of cancer is not always reflected in the appearance of its most common cellular component. Mature glial cells divide in the adult brain, suggesting that they are susceptible to neoplastic transformation. However, another proliferative population of cells (called neural stem cells) has been discovered in the adult brain of mouse and human, indicating that they are much more vulnerable to oncogenic transformation [5,6]. Thus, the classic hypothesis of glioma initiation must be reevaluated to obtain additional insight into the origin of glioma. In this review, we discuss the role of brain tumor stem cells (BTSCs, also known brain tumor-initiating cells), which are currently thought to be a major cause of brain tumor initiation and recurrence [7,8].
Go to : 
BRAIN TUMOR STEM CELL MICROENVIRONMENTS
Various adult stem cells localize within protective and supporting microenvironments (also known as niches) that are composed of a number of differentiated cells [9]. These differentiated cells are known to participate in direct cell-cell contacts and provide paracrine factors, such as growth factors or cytokines, to primarily sustain stem cell traits in a relatively quiescent state. For instance, neural stem cells and BTSCs exist within a vascular niche in which endothelial cells regulate stem cell self-renewal [10-13]. Thus, it is widely assumed that BTSCs arise from neural stem or progenitor cells that are acquired through various genetic and epigenetic alterations, enabling them to undergo neoplastic transformation and escape from vascular niche control [14-16]. Alternatively, uncontrolled proliferation and transformation of neural stem cells may occur by deregulation of external signaling factors within the vascular niche [17]. Therefore, if BTSCs depend on aberrant microenvironments, these niche microenvironments may represent targets for cancer treatment (Fig. 1).
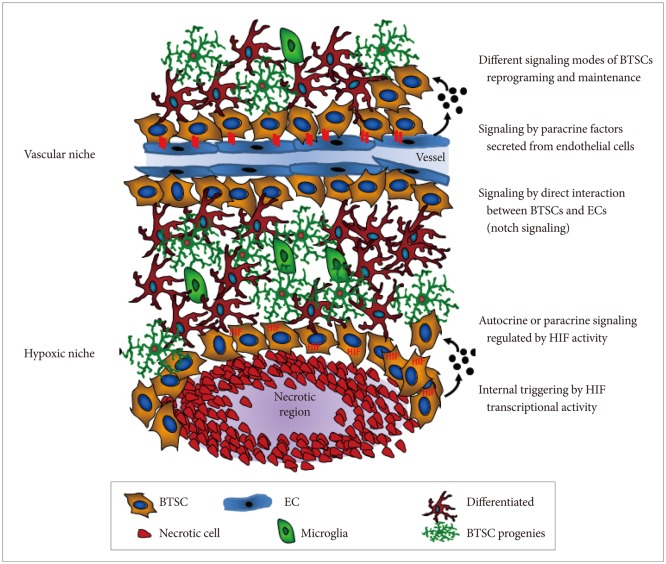
Nitric oxide synthases (NOS) are a family of enzymes that produce nitric oxide (NO) from their substrate, L-arginine. NO regulates many physiological processes through the NO/cyclic guanosine monophosphate (cGMP) pathway and protein S-nitrosylation [18]. For NO signaling, NO diffuses to cells in which it binds to its cytoplasmic receptor, soluble guanylyl cyclase (sGC) that generates cGMP from GTP and subsequently activates several downstream molecules, such as cGMP-dependent protein kinase (PKG). The endothelial isoform of NOS (eNOS) is elevated in various cancer types [19], including human gliomas, where its expression is correlated with glioma grade [20]. Increased eNOS activity in glioma is observed in tumor endothelial cells [20]. Additionally, constitutive NOSs (nNOS and eNOS) are up-regulated, while inducible NOS (iNOS) are down-regulated in primary brain tumors [21]. It is also known that NO exerts paradoxical effects on a cell; it can confer resistance to apoptosis and enhance proliferation, but also can be toxic by inducing DNA damage responses and inhibiting electron transport [22,23]. Apoptosis resistance and proliferation enhancement are attributable to cNOS, which is found in excess in high-grade tumors, while the toxicity appears to be regulated by micromolar levels of NO induced by iNOS, which is most often associated with a host immune response. Recent studies have shown that NO produced in endothelial cells stimulates self-renewal of BTSCs that are enriched and interact with the vasculature by activating NOTCH signaling [20,24]. However, the specific roles of NO and NOS in controlling BTSCs in the vascular niche has not been fully established.
The inhibitor of differentiation (ID) family includes 4 members of the helix-loop-helix (HLH) proteins (ID1-4) that lack a DNA-binding domain, interact with basic HLH (bHLH) transcriptional factors, and function as negative regulators of differentiation during development [25]. ID proteins promote cell proliferation by suppressing cell-cycle-negative regulators, such as p21WAF1/Cip1 and p16INK4a [26,27]. ID1 and ID3 are required for tumor angiogenesis and secondary breast tumor re-initiation in lung metastasis [28,29]. Loss of ID1 and ID3 results in severe defects during vascular development in embryonic mouse brains with massive hemorrhage [30]. Overexpression of ID proteins leads to transdifferentiation of fibroblasts into neural stem-like cells, presumably due to its prominent role in governing neural stem cell fate [31]. ID proteins are also involved in controlling the self-renewal activity of induced pluripotent stem cells (iPSCs) in combination with reprograming factors such as OCT3/4, SOX2, and NANOG [32]. Thus, ID proteins may be involved in regulating the vascular niche and stem cell self-renewal. We recently showed that ID4 induces the genesis of BTSCs through NOTCH-dependent dedifferentiation of Ink4a/Arf-deficient mouse astrocytes [33], while epidermal growth factor receptor signaling induces genesis of BTSCs and angiogenesis through ID1/ID3-dependent activation of cytokines, such as interleukin (IL)-6, IL-8, and GRO1 [34]. Transforming growth factor-β signaling induces expression of ID1 and ID3 to regulate the tumor-initiating capacity of BTSCs [35]. Thus, understanding the precise roles of ID proteins may facilitate the development of effective BTSC-targeting therapeutic agents.
Increasing evidence suggests that embryonic stem cells (ESCs) are exposed to low oxygen levels (hypoxic microenvironment) due to the absence of vasculature in early embryos [36] and that adult stem cells are localized in specific niches characterized by a hypoxic microenvironment [37]. Because stem cells are the only cell type that is not replaced during the entire life span of animal, it is likely that the hypoxic niche protects stem cells from harmful damage induced by oxygen or other reactive oxygen species (ROS) [38]. A number of studies in culture have demonstrated a direct relationship between oxygen levels and stem cell proliferation, differentiation, and survival [39]. Hypoxia leads to several changes in gene expression triggered by a group of transcription factors known as hypoxia-inducible factors (HIFs). These factors belong to the bHLH and PER-ARNT-SIM families and are composed of 2 subunits: alpha subunits (HIF-1α, -2α, or -3α) are variable and oxygen sensitive, but rapidly stabilized in response to low oxygen levels; HIF-1β, also known as aryl hydrocarbon receptor nuclear translocator, is constant and constitutively expressed [40,41]. When a cell is exposed to low oxygen conditions, the α subunit is stabilized and binds to the β subunit; the resulting complex translocates into the nucleus. There, the complex activates transcription of specific genes by recognizing promoter regions known as hypoxia-responsible elements. Increased HIF-regulated genes mediate a number of changes at both cellular and systemic levels. The relationship of hypoxia with tumor progression has been reported in clinical studies in which patients with hypoxic tumor showed a significantly poor clinical outcome [42,43]. Hypoxia correlates with increased tumor invasion and metastasis, as well as higher resistance to radiotherapy and chemotherapy [44]. This is due in part to hypoxia-dependent expression of drug-resistance genes [45], selection of apoptosis-resistant clones [46], and disruption of DNA repair mechanisms [47]. As shown in Fig. 1, cancer stem cells (CSCs) are also located in low-oxygen regions, a key feature of the stem cell microenvironment. Hypoxia may regulate stem cell localization and maintenance by inducing expression of paracrine factors in an HIF-dependent manner. Therefore, hypoxia may maintain the undifferentiated state of CSCs and thus contribute to cancer growth, invasion, and metastasis. Surprisingly, BTSCs preferentially locate near the tumor vasculature and interact with tumor-associated endothelial cells [10]. Although this observation may partially contradict the hypoxic niche hypothesis of CSCs, in vitro co-culture experiments showed that paracrine factors secreted by endothelial cells promote CSC growth, self-renewal, and proliferation. Given that CSCs require both vasculature and hypoxia, the role of endothelial cells within the CSCs niche may be independent of their vascular function, such as supplying oxygen. Indeed, this assumption is supported by the presence of endothelial precursors without vascular functionality near tumor cells and the abnormality of tumor-associated blood vessels that give rise to hypoxic or anoxic regions in solid tumors.
Go to :
REPROGRAMING OF BRAIN TUMOR CELLS INTO BRAIN TUMOR STEM CELLS
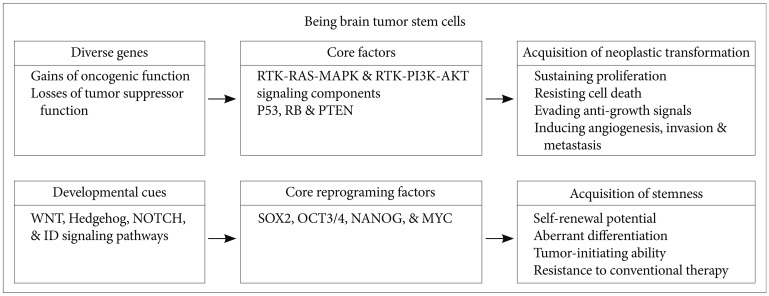
A recent seminal study demonstrated that 4 transcription factors (known as reprogramming factors), OCT3/4, SOX2, c-Myc, and Klf4, convert embryonic and adult fibroblasts to pluripotent cells, known as iPSCs [48]. This is a proof of principle that pluripotent stem cells can be generated from somatic cells through the combination of a small number of factors that rewind the clock of unipotent cells and convert them to a state resembling ESCs.
Interestingly, these reprogramming factors also appear to be up-regulated, either alone or together, in the CSCs of various solid tumors, including brain tumor [49,50]. The current CSC hypothesis involves the existence of a small population of tumor cells possessing unique self-renewal and tumor-initiating capabilities, which differs from other tumor cells [51]. In support of this hypothesis, CSCs have been identified in different cancer types, such as GBM, breast cancer, prostate cancer, colon cancer, and leukemia [52-57]. Numerous BTSC markers exist, including CD133, CD15, and A2B5 membrane markers [58,59], Nestin filament marker [60,61], Musashi-1 RNA-binding protein [62], and OCT3/4, SOX2, ID1, ID3, ID4, IRF7, and BMI1 transcription factors [34,63-65].
Two of these reprogramming factors, OCT3/4 and SOX2, show increased expression levels in patients with glioma. Expression of OCT3/4, a well-known regulator of self-renewal and differentiation in ESCs, is reactivated during conversion of normal cells into neoplastic cells [66]. Similarly, several recent studies have suggested that adult stem cells expressing the OCT3/4 gene initiate the carcinogenic process [67,68]. Moreover, some recent studies have demonstrated that OCT3/4 is overexpressed in various human malignancies, including gliomas, bladder carcinoma, lung adenocarcinoma, ovarian carcinoma, and testis tumors [63,69]. Indeed, overexpression of OCT3/4 can lead to epithelial dysplasias by blocking differentiation of progenitor cells [70]. Increased SOX2 expression has been reported in a growing list of tumors, including GBM, medulloblastoma, breast cancer, prostate cancer, and lung cancer [71-73]. SOX2 expression in brain tumors is not surprising, because SOX2 is mainly expressed in neural stem and progenitor cells of normal brain [74]. In BTSCs and GBM patient samples, hypomethylation of the SOX2 promoter directly correlates with its expression levels [75]. Additionally, GBM has the SOX2 gene amplification (~10%) and overexpression (>80%) [70]. Moreover, knockdown of SOX2 decreases the proliferation and tumorigenicity of GBM [76]. Ectopic expression of SOX2 allows glioma cells to acquire self-renewal and aggressive tumor-initiating abilities as well as resistance to chemotherapy by inducing the expression of multidrug resistance genes, including ABCC3 and ABCC6 [64]. Thus, as shown in Fig. 2, alterations in the expression of 1 or more reprograming factors may trigger the conversion of a given differentiated cancer cell into a malignant cell with self-renewal, aberrant differentiation, and tumor-initiating abilities, as well as resistance to current standard therapy. It is also possible that the tumor microenvironment leads to genetic and epigenetic changes that activate reprograming factors, thereby giving rise to the conversion of glioma cells into BTSCs.
Go to :
CONCLUSION
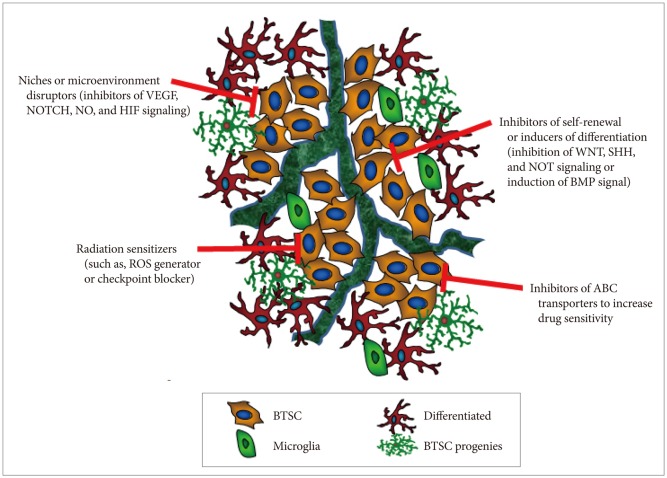
Most conventional cancer therapies are directed to target rapidly dividing cells, which represent most of the tumor cell population. However, in many cases, these therapies fail to eliminate the stem-like cell fraction of the tumor, leading to tumor relapse and selection of more aggressive, therapy-refractory cancer cells. Development of therapeutic modalities specifically targeting BTSCs appear to be necessary in order to achieve complete tumor remission and prevent tumor recurrence after the therapy. The use of patient-derived tumor spheroids or the more aggressive, therapy-resistant tumor cells are valuable for identifying varying properties of BTSCs through transcriptomes and proteomics analyses. Several strategies can be used to develop molecular tailored therapeutic agents and selectively eliminate BTSCs, which are highly resistant to radiotherapy and chemotherapy (Fig. 3): 1) self-renewal inhibitors or differentiation inducers that target various external or internal stemness signaling cues, such as the WNT, SHH, and NOTCH pathways; 2) BTSC niche and microenvironment disruptors, such as inhibitors of VEGF, NO, and HIF signaling; 3) inhibitors of ABC transporters (multidrug resistance proteins) that increase the efficacy of conventional chemotherapeutic agents; and 4) radiotherapy sensitizers, such as ROS generator and checkpoint blocker. Therefore, based on our current understanding of BTSC biology, development of novel therapies specifically targeting BTSCs may be effective for treating essentially incurable malignant tumors in the central nervous system.
Go to :
 XML Download
XML Download